Original Data
| Rev Diabet Stud,
2007,
4(3):147-158 |
DOI 10.1900/RDS.2007.4.147 |
Expression Analysis of cPLA2 Alpha Interacting TIP60 in Diabetic KKAy and Non-Diabetic C57BL Wild-Type Mice: No Impact of Transient and Stable TIP60 Overexpression on Glucose-Stimulated Insulin Secretion in Pancreatic Beta-Cells
Iver Nordentoft1, Per B. Jeppesen1, Anders L. Nielsen2, Poul Jorgensen3, Kjeld Hermansen1
1Department of Endocrinology and Metabolism C, Aarhus Sygehus THG, Aarhus University Hospital, Tage-Hansens Gade 2, DK-8000 Aarhus C, Denmark
2Institute of Human Genetics, The Bartholin Building, University of Aarhus, DK-8000 Aarhus C, Denmark
3Department of Molecular Biology, Aarhus University, C.F. Møllers Allé 130, DK-8000 Aarhus C, Denmark
Address correspondence to: Iver Nordentoft, e-mail: iver.nordentoft@ki.au.dk
Keywords: type 2 diabetes, beta-cell, TIP60, MIN6 cell, INS-1E cell, gene expression
Abstract
In the present study we investigate the expression levels of cytosolic phospholipase A2 α (cPLA2α) interacting histone acetyl transferase proteins TIP60α and TIP60β in non-diabetic C57BL wild-type mice and obese type 2 diabetic KKAy model mice. The aim was to test our hypothesis that TIP60 plays a regulatory role in glucose-stimulated insulin secretion from pancreatic β-cells. MATERIAL AND METHODS: Ten obese diabetic KKAy mice and ten non-diabetic C57BL mice were fed a standard chow diet. After nine weeks, islet RNA was purified and used to measure TIP60 expression. We investigated the effect of TIP60α and TIP60β on glucose-stimulated insulin secretion by transient and stable overexpression in the pancreatic mouse β-cell line MIN6 and the rat β-cell line INS-1E. RESULTS: We found that non-diabetic C57BL mice and diabetic KKAy mice have the same level of both the α and β splice forms of TIP60. Furthermore, we demonstrated that transient and stable expression of TIP60 in INS-1E cells affects neither glucose-stimulated insulin secretion, insulin output nor cell insulin content. Also susceptibility to developing gluco-toxicity was unaffected. CONCLUSION: TIP60 over-expression does not affect glucose stimulated insulin secretion, insulin content or abnormal β-cell function during glucotoxicity.
Introduction
To understand the cellular and molecular mechanisms responsible for type 2 diabetes (T2DM) it is necessary to conceptualize the framework within which glycemia is controlled. Insulin is the key hormone for regulation, and normoglycemia is maintained by the balanced interplay between insulin secretion and insulin action. There is little doubt that transcriptional regulatory factors have an important function in T2DM. For example, the peroxisome proliferator-activated receptor γ (PPARγ) plays a pivotal role in insulin resistance in adipose tissue [1]. PPARγ is activated by certain fatty acids, prostanoids and thiazolidinediones [1]. In β-cells, other trans-cription factors e.g. the peroxisome proliferator-activated receptor-γ coactivator 1α (PGC1α), play a regulatory role. Overexpression of PGC1α in β-cells decreases the levels of a number of glycolytic enzymes, causing glucose intolerance after islet transplantation in vivo [2].
Many authors suggest that the long-chain polyunsaturated fatty acid arachidonic acid (AA), which constitutes at least 30% of the fatty acyl content of islet glycerophospholipids, is critically involved in the stimulatory regulation of insulin secretion from pancreatic β-cells, either as intact AA or as biologically active metabolites generated by the action of oxygenase enzymes [3, 4].
Interestingly, during serum deprivation, the transcription factor TIP60β co-localizes with cytosolic phospolipase A2 (cPLA2) in the nucleus and potentiates cPLA2-mediated prostaglandin E2 (PGE2) production in rat mesangial cells for example in response to serum deprivation [5]. The group IV cytosolic phospholipase A2 (cPLA2 α) hydrolyzes phospholipids to free fatty acids (FFA) in the cellular membranes and has selectivity for diacylphospholipids containing AA at the sn-2 position, thereby liberating AA, the precursor of PGE2 [6]. Interestingly, cPLA2 α was found to interact directly with the transcription factor TIP60 (also named cPLA2-interacting protein, PLIP) [5]. TIP60 is a histone acetyltransferase (HAT) protein belonging to the MYST family, named after its founding members: MOZ, Ybf2/Sas3, Sas2 and TIP60. TIP60 is present in both nucleus and cytoplasm. Two TIP60 protein isoforms, TIP60α and TIP60β exist as a result of alternative mRNA splicing. Both of the TIP60 isoforms contain the cPLA2 α interacting domain. Homologues of TIP60 from different species share considerable homology with human TIP60. The amino acid sequence of TIP60α from human, mouse and rat diverges only at position 175. The mouse and rat sequences are identical and both have a serine at position 175 instead of the alanine found in the human protein. Since the 52 amino acid encoded by exon 5, which is excluded from the β isoform, consists of amino acids 96-147, the alanine to serine substitution is also present in the β homologues.
The function of TIP60 in β-cells is potentially very interesting with regard to the interaction with cPLA2 that generates AA on subsequent stimulation of insulin secretion. However, AA is also the precursor for PGE2 that inhibits insulin secretion. Islet cPLA2 activation is increased in insulin-resistant mice [7]. Furthermore, TIP60 interacts with the ETA receptors in various tissues and we have previously demonstrated that ET-1 influences insulin secretion [8, 9].
According to these observations, we hypothesize that Tip60 could influence β-cell functions and thereby insulin sensitivity and the propensity to develop T2DM. T2DM and the insulin resistance syndrome are increasing in prevalence with alarming rapidity as a result of life-style changes against a background of genetic susceptibility. To develop effective future strategies for treating T2DM, the unraveling of its pathophysiology is fundamental, including the molecular link to insulin resistance. In addition, dysfunction of the insulin-producing β-cell of the pancreas during development of insulin resistance is recognized as a major reason for the escalation of peripheral insulin resistance and progression to overt T2DM.
In the present study, we investigated the expression levels of TIP60α and TIP60β in healthy non-diabetic C57BL wild-type mice and the T2DM KKAy model mice. We also monitored the effect of transient and stable overexpression of TIP60α and TIP60β isoforms in the pancreatic mouse β-cell line MIN6 and the rat β-cell line INS-1E.
Materials and methods
Animals
Ten male KKAy-mice (Clea Japan, Tokyo, Japan), all 5 weeks old, weighing 22-25 g, were fed a standard chow diet. As a non-diabetic control group, ten normal C57BL mice (Taconic, Ry, Denmark) were fed with standard chow diet. The composition of the standard chow diet dry matter was as follows: protein 24%, carbohydrate 71% and lipids 5%. We used 12-hour light/dark cycles. The Danish Council for Animal experiments has approved the study.
Plasma analysis
Blood samples, taken from the tails of the animals, were placed into chilled tubes containing heparin/aprotinin and centrifuged (4000 × g, 60 seconds, 48°C). The plasma was frozen for subsequent analysis of insulin and glucose content. Fasting blood glucose was measured before and after intervention. Plasma blood glucose was determined using the glucose oxidase method (GOD-PAP, Boehringer Mannheim, GmbH Germany).
Insulin assays
Insulin was determined by radioimmunoassay (RIA) using a guinea pig antiporcine insulin antibody (PNILGP4, Novo Nordisk, Bagsvaerd, Denmark), mono-[125I]-(Tyr A14) labeled human insulin (Novo Nordisk) as tracer and rat insulin (Novo Nordisk) as standard. We separated free and bound radioactivity using ethanol [10]. Assays were considered to have been successfully performed when interassay and intra-assay variations were below 10%. The insulin concentration was adjusted to the cell number.
To measure insulin secretion, cells were pre-incubated in a modified Krebs-Ringer buffer (125 mM NaCl, 5.9 mM KCl, 1.28 mM CaCl2, 5.0 mM NaHCO3, and 25 mM HEPES, pH 7.4, all from Sigma) containing 3.3 mM glucose and 0.1% BSA at 37°C for 30 min. After pre-incubation, glucose-stimulated insulin secretion was determined by incubating the cells in either 3.3 and 16.7 mM or 2.0 and 15.0 mM glucose for 60 min. After incubation, 300 μl medium was collected and centrifuged. 200 μl aliquot supernatant was subsequently stored at -20°C for insulin assays (RIA).
To measure insulin output, 100 μl culture medium (from 24-well plates) was transferred to Eppendorf tubes and frozen at -20°C for later assay (RIA).
To measure insulin content 48 h post transfection, or 72 h post cell seeding of stably transduced cell lines, the medium was replaced by 1 ml glycine-BSA buffer (100 mM glycine and 0.25% BSA, pH 8.8) and cells were detached using a rubber policeman and transferred to Eppendorf tubes. Insulin was subsequently released by sonication (Sonifier 250, Branson, Danbury, CT) on ice for 15 sec (twice), each time. After centrifugation for 30 min at 16,000 rpm, the supernatant was collected and frozen at -20°C for later analysis. Insulin content was adjusted to cell number.
Expression Plasmids
Plasmids of mouse TIP60 p3xFLAG-CMV 7.1-TIP60α, p3xFLAG-CMV 7.1-TIP60β and pEGFPC1-TIP60β were generously provided by Dr. Xanthi Merlo, Medical College of Wisconsin, Milwaukee, WI, USA. pEGFPC1-Tip60α was generated by subcloning from p3xFLAG-CMV 7.1-TIP60 into the pEGFPC1 vector (Clontech). Retroviral expression vector pBabe-puro, encoding mouse Tip60α (gene bank AF528195), was generated by sub-cloning from pCDNA4/TO/myc-His-Tip60α using BamHI/PmeI. Tip60β (gene bank AF528196). Prior to sub-cloning, TIP60α and TIP60β were PCR cloned (EcoRI/NotI) from p3xFLAG-CMV 7.1-TIP60α and p3xFLAG-CMV 7.1-TIP60β using primer pairs 5'-CGCGAATTCAGCAATGGCG-3', 5'-TTTGCGGCCGCCACTTT-CCTCTCTTGCT-CC-3' and 5'-TATGAATTCAATATGGCGGAGGTGGGGA-3', 5'-TTTGCGG-CCGCCACTTTCC-TCTCTTGCTCC-3' respectively, into pCDNA4/TO/myc-His (Invitrogen). TIP60 clone pBabe-puro-Tip60β was generated from pBabe-puro-Tip60α by excision of exon 5 using quick change site-directed mutagenesis with primer QCTIPalpha→beta(+) 5'-CCCGAAAGAGAGGT-GAAACGGAAGGTG-GAGG-3' and QCTIPalpha→beta(-) 5'-CCTCCACCTTCCGTTTCACCTCTCTTTC-GGG-3' using Pfu ultra HF DNA polymerase (Stratagene). All cloned inserts were verified by sequencing (ABI 3100, ABI).
Cell Culture
Insulinoma MIN6 cells (generously donated by Dr Yamamoto, Kumamoto University School of Medicine, Japan) were maintained in Dulbeccos’s modified Eagle's medium (DMEM) containing 25 mmol/l glucose, 1% penicillin/strepto-mycin, 10% heat-inactivated fetal calf serum (FCS) and were cultured at 37°C with 5% CO2. Cells were refed every 3-4 days and passaged every 7 days. The passage number of INS-1E cells (provided by Prof. C. B. Wolheim, Geneva, Switzerland) used in this study was 65-80. INS-1 cells were maintained at 11.1 mM glucose in a modified RPMI 1640 medium (see below). The INS-1E cells were grown as previously described [11]. Simultaneously, cells were subcultured for 72 h in 24-well plates for secretion studies.
Western blotting
INS-1E cells transiently transfected at 60% confluence (6-well plates) and cultured for 48 h as well as stable TIP60-expressing INS-1E, cultured with puromycin 1 μg/ml to 80% confluence in 6-well plates, were used to make protein lysate. Whole cell protein lysate was collected by centrifugation after lysis with cold RIPA (RadioImmunoPrecipitation Assay) buffer (sc.24948, Santa Cruz, CA, USA) supplemented with complete Protease Inhibitor Cocktail (Roche). Protein concentration was determined using BioRad detergent-compatible protein kit (BioRad laboratories, Hercules CA, USA). A 25 μg protein sample was resolved by SDS-PAGE (CriterionTM 4-20% Tris-HCL; BioRad). Proteins were transferred to PVDF membrane (Amersham International, UK) and blocked overnight with 5% non-fat milk in TBS. Membranes was probed with primary anti-TIP60 rabbit polyclonal IgG (2 μg/ml UpState) or anti-actin mouse monoclonal IgG (A5441, 1:5000, Sigma) and, subsequently, secondary horseradish peroxidase-conjugated goat anti-rabbit immunoglobulins (Sc-2004, Santa Cruz 1:2000) or bovine anti-mouse (Sc-2375, Santa Cruz 1:5000). Bands were visualized using enhanced chemoluminescence (ECL, Amersham International, UK).
Cell count
Cells in 24-well plates were washed with PBS (room temperature) and 1 ml PBS was added to each well. The background was measured on FluoroStar (excitation 490 nm and emission 520 nm) at gain 60. 20 μl of SYTO 24 (Molecular Probes S-7559, 5 mM diluted 1:500 in DMSO) were then added to each well and incubated for 30 min at 37°C, followed by measurement. Relative cell numbers were calculated by subtracting the background values.
Glucotoxicity
INS-1E cells were seeded in 24-well plates at 2 × 105 cells/well. After 24-hour culture, the medium was removed and replaced with culture medium containing 11.1 mM, 16.7 mM, 20.0 mM or 25.0 mM glucose and cultured for 48 h. Subsequently, we washed the cells in UB buffer for 15 min and measured glucose-stimulated insulin secretion (GSIS) in response to 2 and 15 mM glucose.
Islet isolation
Islets were isolated as previously described [9].
Isolation of RNA
INS-1E and MIN6 RNA was isolated from 80% confluence 6-well and 24-well plates using RNeasy spin columns (Qiagen, Hilden, Germany), as recommended by the manufacturer. RNA from mice islets were purified using Trizol reagent (Gibco/Invitrogene, Calsbad, CA, USA). For each group, islets from 3-4 mice (180-200 islets) were pooled in 1 ml Trizol reagent before RNA purification. Total RNA was extracted according to the manufacturer’s instructions. RNA was quantified by measuring absorbance at 260 and 280 nm.
Real-time RT-PCR
We investigated the expression of TIP60α and, TIP60β by real-time RT-PCR. cDNA was synthesized using IScript (BioRad, Hercules, CA, USA), according to the manufacturer’s instructions. In the transient transfection experiments, the RNA preparation was treated with TURBO DNA-free DNase (Ambion, Austin, TX, USA) prior to cDNA synthesis. Total RNA at 50 ng per 10 μl of reaction mixture was used for measurement of the target mRNA. The real-time RT-PCR assay was performed using an ABI 7500 FAST machine (ABI, Foster City, CA). The 10 μl real-time RT-PCR reagents consisted of 5 μl 2 × TaqMan® FAST Universal Master Mix (P/N 43660783, ABI, Foster City, CA), 0.5 μl 20 × TaqMan® Assay/probe (ABI, Foster City, CA) and cDNA equivalent to 50 ng of total RNA in 4.5 μl H2O. The thermal FAST cycle program was as follows: 20 s at 95°C followed by 40 cycles of 3 seconds at 95°C and 30 seconds at 60°C. Reactions were set up in triplicate for each sample and expression of genes under analysis was normalized to eukaryotic 18S rRNA expression (assay Hs99999901_s1, ABI, Foster City, CA). All assays were performed in 96-well format plates covered with optical adhesive cover (P/N 4346906 and P/N 4311971 ABI, Foster City, CA). We used the 2-ΔΔCT method to calculate relative gene expression. The TaqMan® assays used were as follows: TIP60α (assay Mm01231511_g1 (mouse)), TIP60β (assay mTIP60b (designed using GeneBuilder (ABI) (mouse)), TIP60α (assay Rn01231511_g1 (rat)) and TIP60β (assay Rn00437443_m1 (rat)).
Transient transfection
The day prior to transfection, 2 × 105 MIN6 or INS-1E cells were plated into 24-well plates in 500 μl culture medium (see cell culture). Mix A: for each well, 1 μg pEGFPC1, pEGFPC1-TIP60α or pEGFPC1-TIP60β was mixed in 50 μl serum-free medium. Mix B: 3 μl TransFectin reagent (BioRad, Hercules, CA, USA) and 50 μl serum-free medium were mixed. Finally A and B were mixed, incubated for 20 min at room temperature and added in a drop-wise manner to each well. Cells were incubated for 48 h and then monitored under fluorescence microscope before conducting the assay for glucose-stimulated insulin secretion. Each plasmid was transfected in 24 wells, allowing 12 wells for low and high glucose stimulation.
Retroviral Transduction
Phoenix packaging cells at 70-80% confluence in DMEM containing 10% FCS in 100 mm culture plates were transfected with 15 μg pBabe-puro-TIP60α and TIP60β plasmid using 50 μl TransFectin (Bio Rad laboratories), according to the manufacturer’s instructions. After 48 h incubation (37°C, 5% CO2), the virus-containing supernatant was filtered through a 0.45 μm sterile filter and immediately used for INS-1E cell transduction. INS-1E cells at 30-40% confluence in 100 mm dishes were transduced by adding a 1:1 mixture of viral supernatant and RPMI supplemented with polybrene to a final concentration of 6 μg/ml. After 24 h, the cells were split 1:3 to avoid cells reaching confluency and were plated in RPMI-1640 containing 11.1 mM glucose + 1 μg/ml puromycin. Cells were selected using 1 μg/ml puromycin. Puromycin-containing medium was changed every second day. Stable transduction was accomplished after 8-10 days compared to untransducted controls. Stably transducted clones were pooled and used for further experiments.
Statistical analysis
Data are expressed as mean ± standard error of the mean (SEM). Statistical significance between the two groups was evaluated using a two-tailed t test. A p-value of less than 0.05 was considered statistically significant. For gene expression analysis, one-way ANOVA was applied.
Results
Mouse models for metabolic effects
In order to investigate if there is a difference in the expression levels of TIP60α and TIP60β in non-diabetic and diabetic islet cells we set up an animal study. Ten non-diabetic wild-type C57BL mice and ten KKAy mice (T2DM model mice), all aged 5 weeks, were fed standard chow diet for 9 weeks. At the start of the intervention study, no significant difference was found in blood glucose or insulin levels between the two groups (Figures 1A and 1B).
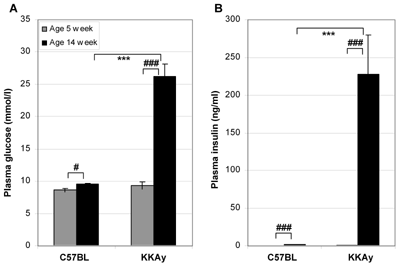 |
 |
Figure 1. Fasting plasma glucose (A) and insulin (B) in KKAy and C57BL mice before and after 9 weeks of feeding. The C57BL and KKAy control groups were fed standard chow diet. The animals were 5 weeks old when treatment began. Data are shown as mean ± SEM (n = 10 in each group). ***p < 0.001 (unpaired), #p < 0.05 (paired), ###p < 0.001 (paired). |
|
After the 9-week feeding period, the KKAy control group developed a significant increase in plasma glucose of 181% (9.4 vs. 26.3 mmol/l, p < 0.001), whereas the non-diabetic C57BL control group only experienced an increase of 10% (8.6 vs. 9.5 mmol/l, p = 0.013). Plasma insulin increased correspondingly i.e. 266-fold (0.9 vs. 234.1 ng/ml p < 0.001) and 4.5-fold (0.4 vs. 1.8 ng/ml p < 0.001) for KKAy and C57BL respectively. Plasma glucose and insulin in the KKAy group were 2.8-fold (9.5 vs. 26.3 mmol/l, p < 0.001) and 130-fold (1.8 vs. 234.1 ng/ml, p < 0.001) higher respectively, compared to the C57BL control group (Figures 1A and 1B).
TIP60 gene expression analysis in C57BL and KKAy mouse islets by real time RT-PCR
To analyze islets from the pancreas of the non-diabetic C57BL mice and the diabetic KKAy mice, RNA was isolated and subsequently analyzed for the expression of TIP60α and TIP60β using real-time RT-PCR (Figure 2). The change in transcript abundance was calculated for the KKAy group compared to the C57BL control group (Figures 2A and 2B). This analysis showed no difference in TIP60α and TIP60β expression between the non-diabetic and the diabetic mice. The TaqMan assays used to detect TIP60α and TIP60β are designed to span exon 4-5 and 4-6 respectively, and thus specifically detected each splice form. Primer efficiency was comparable. On the assumption that reverse transcription for the two mRNA is comparable, the qPCR analysis shows that the abundance of TIP60β mRNA is approximately 3-fold higher than TIP60α mRNA in both the C57BL mice and the KKAy mice (Figure 2C). To investigate the expression level of TIP60 in islets, we purified RNA from liver and muscles from C57BL mice and compared the expression in islets and MIN6 cells. Both TIP60α and TIP60β expression in islets are comparable to that in muscles, but higher than that in liver (Figures 2D and 2E). The expression levels of TIP60α and TIP60β in clonal MIN6 cells were 5-fold higher than in the islets, indicating that TIP60 is relatively highly expressed in β-cells (Figures 2D and 2E).
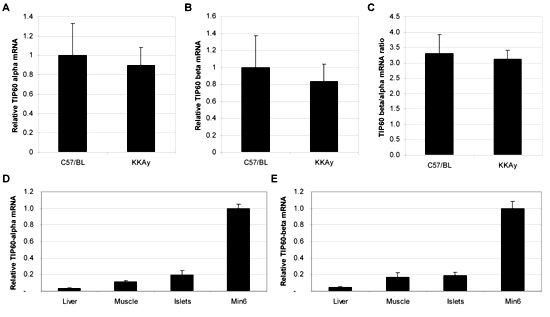 |
|
Figure 2. RNA from pancreatic islets of C57BL and KKAy mice fed for 9 weeks with chow was isolated and the transcript abundance of TIP60α and TIP60β mRNA was measured using real-time RT-PCR. A: relative TIP60α expression. B: relative TIP60β expression. C: ratio of TIP60β mRNA and TIP60α mRNA. To evaluate TIP60 expression levels across tissues, RNA was prepared from liver and muscles from the C57BL group and the comparative transcript abundance of TIP60α (D) and TIP60β (E) mRNA in liver, muscles and MIN6 cells was measured. Measurements were carried out in triplicate for each sample and gene expressions were normalized to 18S rRNA expression (n = 4). |
|
We also measured the expression of insulin 1 and 2 (Ins1/2), the central β-cell transcription factors Pdx1/Ipf1 and beta2/neurod1 regulating insulin expression (Figure 3). No difference was detected in Ins1/2 expression between the C57BL mice and KKAy mice. Similarly, no changes were observed for Pdx1 and beta2 (Figure 3). Furthermore, we measured the expression of the insulin receptor and glucose transporter GLUT2. GLUT2 expression was 2.5-fold lower in KKAy mice than in C57BL mice.
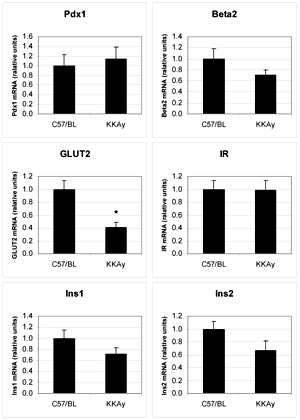 |
|
Figure 3. RNA from pancreatic islets of C57BL and KKAy mice fed for 9 weeks with chow was isolated and genes related to insulin secretion and regulation were studied for changes in their transcript abundance (TA) by real-time RT-PCR using TaqMan assays. Measurements were carried out in triplicate for each sample and gene expressions were normalized to 18S rRNA expression. Changes in TA were calculated for the KKAy group compared to the C57BL control group. *p < 0.05 (n = 4). |
|
GSIS in MIN6 and INS-1E cells transiently transfected with TIP60α and TIP6β
To investigate the influence of TIP60 overexpression in β cells and any potential difference between the two major splice variants, we used two vector constructs encoding mouse TIP60α or TIP60β fused to EGFP under the control of the CMV promoter (EGFP-TIP60α and EGFP-TIP60β). β-cell lines are known to be notoriously difficult to transfect, so the fluorescence marker EGFP enabled us to monitor transfection efficiency directly by microscopy, easing optimization. We carefully optimized transfecton of MIN6 cells using Fugene (Roche), Superfect (Qiagen), DOTAP (Roche) and Transfectin (Bio-Rad). The transfection efficiency obtained, however, was 40-50%. Moreover, we noted that transfection impaired growth. When we purified RNA 48 h post transfection, extracted RNA followed by DNase treatment and real time RT-PCR, we found TIP60α and TIP60β mRNA levels to be elevated more than 500-fold (data not shown). Results from GSIS analysis 48 h post transfection were very inconsistent from experiment to experiment. We concluded that the low transfection efficiency resulted in more variation, as the high transcription of TIP60 in the successfully transfected cells resulted in toxicity and impaired growth. Previously, TIP60 was ascribed a role in apoptosis [12]. The problems with the MIN6 cell line prompted us to try using the rat β-cell line, INS-1E. After optimization, we obtained 80-100% transfection efficiency. TIP60α and TIP60β mRNA levels measured by real time RT-PCR showed an increase of 1815 ± 147 and 754 ± 117 fold respectively (Figures 4A and 4B). Both TIP60α- and TIP60β-fused EGFP were successfully translated, as evaluated by western blotting and fluorescence microscopy of fixed cells (Figures 4C-D and 4I-J). Cells were in good condition 3 days after transfection, so we proceeded with analysis of glucose-stimulated insulin secretion (Figures 4E-H). As seen from GSIS, performed at passage 70 (upper panel) and passage 74 (lower panel), the INS-1E cells gave a good stimulation at high glucose (16.7 mM) compared to low glucose (3.3 mM). There was, however, no difference in GSIS, glucose output or cell insulin content between the EGFP and EGFP-TIP60α and EGFP-TIP60β-expressing cells (Figures 4E-H).
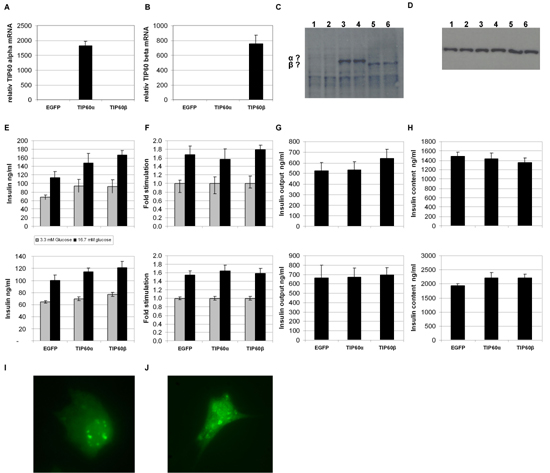 |
|
Figure 4. INS-1E cells were transiently transfected with EGFP-TIP60α and EGFP-TIP60β and empty EGFP vector. The transcript abundance of TIP60α (A) and TIP60β (B) mRNA was measured 48 hours after transfection using real time RT-PCR (n = 4). C: Western blot of 30 µg total protein lysate from EGFP (lane 1-2), EGFP-TIP60α (lane 3-4) and EGFP-TIP60β (lane 5-6) transfected INS-1E cells 48 h post transfection, probed with anti-TIP60 antibody. D: Western blot as figure C probed with actin antibody. E-H: Glucose-stimulated insulin secretion at 2 mM glucose (low) and 15 mM glucose (high) (E), stimulated GSIS going from low to high glucose (F), insulin output from culture medium (G) and cell insulin content (H) for passage 70 (upper panel) and passage 74 (lower panel) (n = 12). I-J: Fluorescence microscopy of fixated EGFP-TIP60α (I) and EGFP-TIP60β (J) transfected INS-1E cells 48 h after transfection. |
|
GSIS in INS-1E cells stably overexpressing TIP60
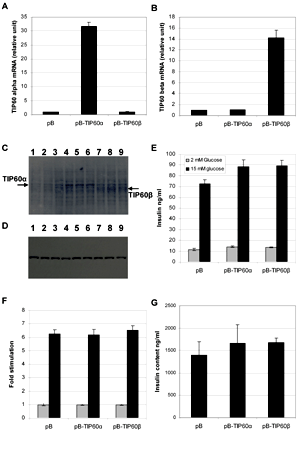
The high expression of TIP60 in the transiently transfected INS-1E cells brings into question the conclusion about the role of TIP60 in GSIS. In order to clarify this, we subcloned TIP60α and TIP60β into the retroviral vector pBabe-retro-puro (pB) and generated stable TIP60-expressing clones of INS-1E, pB-TIP60α and pB-TIP60β. The TIP60 expression of these stable clones was measured by real time RT-PCR using splice specific TaqMan probes. pB-TIP60α- and pB-TIP60β- transduced INS-1E were found to expresses 37.8 ± 1.4, p < 0.01 and 14.2 ± 1.5, p < 0.01 times more TIP60 mRNA, respectively, than a control clone stably transducted with the empty pBabe vector (Figures 5A and 5B). TIP60α was successfully translated (Figures 5C-D), whereas TIP60β was expressed to a lesser extent, in accordance with mRNA levels (Figure 5D). Moreover, TIP60β is more difficult to visualize because of band overlapping with cross-reactive protein. No difference in morphology or growth rate was detected between the stable TIP60 overexpressing INS1 cell lines, the stable empty pB vector INS-1E cells and passage-matched non-transduced INS-1E cells. Therefore, the presence of 14 to 37-fold more TIP60 mRNA does not seems to confer short-term toxicity to the cells. We proceeded to measure glucose-stimulated insulin secretion in the TIP60 expressing INS-1E cells. However, neither TIP60α nor TIP60β expression resulted in significant differences in regulated insulin secretion or insulin content (Figures 5E-G).
 |
|
Figure 5. INS-1E cells were transducted with the following retroviral expression plasmids, pSUPER-retro-puro, pSUPER-TIP60α and pSUPER-TIP60β followed by puromycin selection. The transcript abundance of TIP60α (A) and TIP60β (B) mRNA was measured using real time RT-PCR. (n = 4). C: Western blot of 30 µg total protein lysate from pSUPER (lane 1-3), pSUPER-TIP60α (lane 4-6) and pSUPER-TIP60β (lane 7-9) stably expressing INS-1E cell lines, probed with anti-TIP60 antibody. D: Western blot as in C, probed with actin antibody. Panels E, F and G show glucose-stimulated insulin secretion (E) at 2 mM glucose (low) and 15 mM glucose (high) stimulated GSIS going from low to high glucose (F) and insulin content (G) (n = 12). |
|
Glucotoxicity
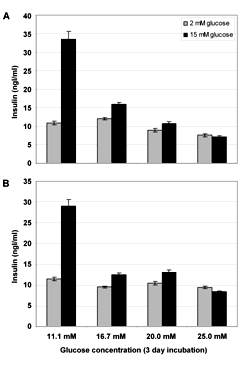
To investigate if TIP60 overexpression changes the β-cells' susceptibility to developing glucotoxicity 24 h post-seeding, stable pB and pB-TIP60α INS-1E cells were changed to medium containing 11.1 mM, 16.7 mM, 20.0 mM or 25.0 mM glucose and cultured for 72 h. Subsequently, we washed the cells in UB buffer and measured GSIS at 2 and 15 mM glucose (Figure 6). As illustrated in Figure 6, 16.7, 20.0 and 25.0 mM glucose pre-incubation rendered the cells unable to secrete insulin; however, at 11.1 mM glucose, both the TIP60α overexpression and the pB control were still able to respond potently to stimulate insulin secretion (approximately 3-fold). TIP60α overexpression, therefore, neither protects nor renders the INS-1E cells more prone to develop glucotoxicity.
 |
|
Figure 6. Glucotoxicity of INS-1E cells stable overexpressing TIP60α. INS-1E stable transduced with empty pBabe (A) or pBabe-TIP60α (B) were seeded in 24-well plates 2 × 105 cells/well. After 24 h, the culture medium was removed and replaced with culture medium containing 11.1 mM, 16.7 mM, 20.0 mM or 25.0 mM glucose and cultured for 48 h. Subsequently, we washed the cells for 15 min. in UB buffer and measured GSIS at 2 and 15 mM glucose Each bar represents the mean ± SEM from 12 incubation wells. * p < 0.05. |
|
Discussion
Insulin resistance and β-cell dysfunction are the primary abnormalities associated T2DM. During the progression of the insulin resistance syndrome, pancreatic β-cells gradually synthesize and secrete more insulin to cope with the increasing insulin requirement and to maintain plasma glucose homeostasis. Consequently, it is essential to clarify gene changes in the β-cells during the development of insulin resistance. The TIP60 protein has been shown to interact with phospholipase A2 (cPLA2) in the cytoplasm and to potentiate cPLA2-mediated prostaglandin E2 production. TIP60 protein belongs to the MYST protein family the members of which all contain the conserved MYST domain that harbors a HAT domain related to the GNAT (Gcn5-related N-acetyltransferase) superfamily [13]. HAT proteins catalyze the post-translational transfer of acetyl group from acetyl coenzyme A to the α-amino group of N-terminal amino acids or the ε-amino group of internal residues. Proteins modified by acetylation include histones and transcription factors. In numerous studies, TIP60 is found to posses a multitude of regulatory functions in relation to gene transcription and it functions as either a co-activator or co-repressor, depending on the context. TIP60 plays a role as a co-activator of nuclear hormone (NR) receptor-mediated transactivation [14] and of the β-amyloid precursor protein Fe65 complex [15]. We have previously shown that TIP60 in HT-1080 cells (human fibrosarcoma cell line) functions as a transcriptional co-repressor for the translocation E26 transforming-specific leukemia (TEL) protein [16]. TIP60 also is demonstrated to co-repress transcription with cAMP-response element-binding protein (CREB) [17] and STAT3 [18]. Accumulating data assign TIP60 an essential role in double-strand break DNA repair through 4 different mechanisms [19-22].
In the present study, we demonstrate that the expression profiles of the α and β isoforms of TIP60 are identical in isolated islets from non-diabetic C57BL mice and from T2DM KKAy mice. The mice were fed standard chow for nine weeks from age 5 to age 14. At age 14, the KKAy mice had developed hyperglycemia and pronounced hyperinsulinemia. We found that the mRNA expression levels of the Ins1 and Ins2 genes were comparable in islets of the KKAy and the C57BL mouse. At first glance this may appear contradictory. However, islets isolated from the KKAy mice were considerably larger in size as was the amount of RNA extracted. This indicates that the β-cell mass of the KKAy mice is increased and β-cell mRNA, including insulin per cell, is up-regulated. However, since the same amount of RNA is always used for relative real time RT-PCR analysis, this difference will be masked.
Adaptation of β-cell mass, primarily by expansion of cell size rather than proliferation, is a common observation during development of insulin resistance to keep up with increased insulin demand and is followed by β-cell apoptosis at the time of overt T2DM. Since the expression of the islet-enriched transcription factors Pdx-1 and beta2 (involved in insulin promoter activation as well as β-cell proliferation [23, 24]) is unchanged, the mRNA ratios seem to be retained. Other transcription factors, e.g. MafA, FOXO, Nkx2.1, Nkx6.1, Pax4, Pax6 and Ngn3, are also critically linked to β-cell growth and development [23, 25, 26], but were not measured in the present study.
The β-cell specific glucose transporter GLUT2 (mouse and rat) is found to be 2.5-fold reduced in KKAy mice, compared to non-diabetic C57BL mice. GLUT2 is the glucose transporter with the lowest affinity and the highest capacity for glucose, allowing β-cells to take up glucose effectively only in times of plentiful glucose, when insulin release is needed. This suggests that the decreased glucose sensitivity observed is partly due to this lower GLUT2 expression [27].
It is important to emphasize that the results obtained with the C57BL control and KKAy mice cannot be extrapolated to other mice models, as strain effect is known to be substantial. Generation of TIP60 β-cell-specific knockout mice using the Cre-loxP technology with Cre recombinase under the control of the insulin promoter could more directly address the effect of TIP60 on GSIS.
To investigate the potential role of TIP60 in glucose-stimulated insulin secretion, we transiently overexpressed the TIP60α and TIP60β isoforms in the clonal pancreatic mouse isulinoma β-cell line MIN-6 and the rat β-cell line INS-1E. We did not achieve conclusive results using MIN-6 cells since we were unable to obtain high transfection efficiency for these cells, despite carefully optimizing with various methods. In addition the cells targeted overexpress TIP60 at very high levels and impaired growth and culture plate detachment occur. The impaired growth was apparently due to cellular toxicity on the part of TIP60, since cells transfected with the empty EGFP vector were not affected. In support of this theory, G418 selection of EGFP-TIP60α transfected MIN-6 cells during 2-week selection lost all EGFP fluorescence in the emerging colonies, indicating selection against TIP60 overexpression. Corroborating this, TIP60 is known to participate in apoptotic signaling [12, 28, 29]. Following this line of evidence, for the strictly controlled amounts of cellular TIP60 to maintain correct cellular functions we note that it has been impossible for us to down-regulate the amount of TIP60 in β-cells by RNAi-based methods. This was performed both by transfection of the cells with an array of different synthetic siRNA duplexes or by transducing cells with retroviral constructs expressing siRNA hairpin precursors.
In contrast, the transfection efficiency of the INS-1E cells consistently exceeded 80% and was not morphologically impaired 48 h. after transfection, indicating that INS-1E cells tolerate TIP60 overexpression (1815 ± 147 fold for TIP60α and 754 ± 117 fold for TIF60β). The results from different passages showed that there was no difference in glucose-stimulated insulin secretion and in glucose output for the TIP60α and TIP60β overexpressing INS-1E cells compared to the control transfected with empty vector. Additionally, using retroviral transduction, we constructed stable INS-1E cells expressing 37 and 14-fold more TIP60α and TIP60β mRNA respectively. These levels are more suited for studying possible physiological changes in the expression of transcription factors that are normally present in small amounts. Again, we did not find any difference in glucose-stimulated insulin secretion or insulin output. Therefore, we conclude that TIP60 overexpression does not regulate GSIS.
During insulin resistance pancreatic islets are challenged by constitutive high plasma glucose levels. This renders β-cells less glucose-sensitive and consequently decreases glucose-stimulated insulin secretion, including first-phase insulin response, developing glucotoxicity. To investigate if TIP60 overexpression changes β-cell function, we tested the stable overexpressing TIP60α INS-1E cell against stable empty vector expressing INS-1E. We found that both cell lines developed the classical glucotoxicity characteristics with a complete loss of glucose responsiveness to high glucose i.e. after incubation with 16.7 mM glucose for 72 h. There was no difference between the cells overexpressing TIP60α compared to the controls, showing that TIP60 neither protects nor enhances the susceptibility for developing glucotoxicity.
In conclusion, TIP60 overexpression does not affect glucose-stimulated insulin secretion, insulin content or the susceptibility of β-cells to develop glucotoxicity and, accordingly, cannot currently be assigned a role in β-cells for glucose-mediated β-cell function in the normal or diabetic state.
Acknowledgments:
We would like to thank Professor Karsten Kristiansen and Rasmus Koefoed Petersen from Department of Biochemistry and Molecular Biology, University of Southern Denmark, for technical support regarding retroviral transduction. Dorthe Rasmussen, Claus Bus, Lene Trudsø, Nanne Højgård, Kirsten Eriksen and Tove Skrumsager are thanked for skilled technical assistance. This study was supported by the Danish Diabetes Association, the Faculty of Health Sciences, Aarhus University, the A.P. Møller Foundation for the Advancement of Medical Science, the VELUX foundation, the Danish Medical Research Council, the Toyota Foundation and the Novo Nordisk Foundation.
References
- Olefsky JM. Treatment of insulin resistance with peroxisome proliferator-activated receptor gamma agonists. J Clin Invest 2000. 106:467-472. [DOD]
- Yoon JC, Xu G, Deeney JT, Yang SN, Rhee J, Puigserver P, Levens AR, Yang R, Zhang CY, Lowell BB, Berggren PO, Newgard CB, Bonner-Weir S, Weir G, Spiegelman BM. Suppression of beta cell energy metabolism and insulin release by PGC-1alpha. Dev Cell 2003. 5:73-83. [DOD] [CrossRef]
- Ahren B, Magrum LJ, Havel PJ, Greene SF, Phinney SD, Johnson PR, Stern JS. Augmented insulinotropic action of arachidonic acid through the lipoxygenase pathway in the obese Zucker rat. Obes Res 2000. 8:475-480. [DOD]
- Persaud SJ, Muller D, Belin VD, Kitsou-Mylona I, Asare-Anane H, Papadimitriou A, Burns CJ, Huang GC, Amiel SA, Jones PM. The role of arachidonic acid and its metabolites in insulin secretion from human islets of langerhans. Diabetes 2007. 56:197-203. [DOD] [CrossRef]
- Sheridan AM, Force T, Yoon HJ, O'Leary E, Choukroun G, Taheri MR, Bonventre JV. PLIP, a novel splice variant of Tip60, interacts with group IV cytosolic phospholipase A(2), induces apoptosis, and potentiates prostaglandin production. Mol Cell Biol 2001. 21:4470-4481. [DOD] [CrossRef]
- Clark JD, Lin LL, Kriz RW, Ramesha CS, Sultzman LA, Lin AY, Milona N, Knopf JL. A novel arachidonic acid-selective cytosolic PLA2 contains a Ca(2+)-dependent translocation domain with homology to PKC and GAP. Cell 1991. 65:1043-1051. [DOD] [CrossRef]
- Simonsson E, Karlsson S, Ahren B. Islet phospholipase A(2) activation is potentiated in insulin resistant mice. Biochem Biophys Res Commun 2000. 272:539-543. [DOD] [CrossRef]
- Brock B, Gregersen S, Kristensen K, Thomsen JL, Buschard K, Kofod H, Hermansen K. The insulinotropic effect of endothelin-1 is mediated by glucagon release from the islet alpha cells. Diabetologia 1999. 42:1302-1307. [DOD] [CrossRef]
- Gregersen S, Thomsen JL, Brock B, Hermansen K. Endothelin-1 stimulates insulin secretion by direct action on the islets of Langerhans in mice. Diabetologia 1996. 39:1030-1035. [DOD] [CrossRef]
- Heding LG. Determination of total serum insulin (IRI) in insulin-treated diabetic patients. Diabetologia 1972. 8:260-266. [DOD] [CrossRef]
- Jeppesen PB, Gregersen S, Poulsen CR, Hermansen K. Stevioside acts directly on pancreatic beta cells to secrete insulin: actions independent of cyclic adenosine monophosphate and adenosine triphosphate-sensitive K+-channel activity. Metabolism 2000. 49:208-214. [DOD] [CrossRef]
- Tang Y, Luo J, Zhang W, Gu W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell 2006. 24:827-839. [DOD] [CrossRef]
- Neuwald AF, Landsman D. GCN5-related histone N-acetyltransferases belong to a diverse superfamily that includes the yeast SPT10 protein. Trends Biochem Sci 1997. 22:154-155. [DOD] [CrossRef]
- Brady ME, Ozanne DM, Gaughan L, Waite I, Cook S, Neal DE, Robson CN. Tip60 is a nuclear hormone receptor coactivator. J Biol Chem 1999. 274:17599-17604. [DOD] [CrossRef]
- Cao X, Sudhof TC. A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science 2001. 293:115-120. [DOD] [CrossRef]
- Nordentoft I, Jorgensen P. The acetyltransferase 60 kDa trans-acting regulatory protein of HIV type 1-interacting protein (Tip60) interacts with the translocation E26 transforming-specific leukaemia gene (TEL) and functions as a transcriptional co-repressor. Biochem J 2003. 374:165-173. [DOD] [CrossRef]
- Gavaravarapu S, Kamine J. Tip60 inhibits activation of CREB protein by protein kinase A. Biochem Biophys Res Commun 2000. 269:758-766. [DOD] [CrossRef]
- Xiao H, Chung J, Kao HY, Yang YC. Tip60 is a co-repressor for STAT3. J Biol Chem 2003. 278:11197-11204. [DOD] [CrossRef]
- Ikura T, Ogryzko VV, Grigoriev M, Groisman R, Wang J, Horikoshi M, Scully R, Qin J, Nakatani Y. Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell 2000. 102:463-473. [DOD] [CrossRef]
- Sun Y, Jiang X, Chen S, Fernandes N, Price BD. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc Natl Acad Sci U S A 2005. 102:13182-13187. [DOD] [CrossRef]
- Murr R, Loizou JI, Yang YG, Cuenin C, Li H, Wang ZQ, Herceg Z. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat Cell Biol 2006. 8:91-99. [DOD] [CrossRef]
- Kusch T, Florens L, Macdonald WH, Swanson SK, Glaser RL, Yates JR, Abmayr SM, Washburn MP, Workman JL. Acetylation by Tip60 is required for selective histone variant exchange at DNA lesions. Science 2004. 306:2084-2087. [DOD] [CrossRef]
- Habener JF, Kemp DM, Thomas MK. Minireview: transcriptional regulation in pancreatic development. Endocrinology 2005. 146:1025-1034. [DOD] [CrossRef]
- Habener JF, Stoffers DA. A newly discovered role of transcription factors involved in pancreas development and the pathogenesis of diabetes mellitus. Proc Assoc Am Physicians 1998. 110:12-21. [DOD]
- Glauser DA, Schlegel W. The emerging role of FOXO transcription factors in pancreatic beta cells. J Endocrinol 2007. 193:195-207. [DOD] [CrossRef]
- Henseleit KD, Nelson SB, Kuhlbrodt K, Hennings JC, Ericson J, Sander M. NKX6 transcription factor activity is required for alpha- and beta-cell development in the pancreas. Development 2005. 132:3139-3149. [DOD] [CrossRef]
- Efrat S. Making sense of glucose sensing. Nat Genet 1997. 17:249-250. [DOD] [CrossRef]
- Sapountzi V, Logan IR, Robson CN. Cellular functions of TIP60. Int J Biochem Cell Biol 2006. 38:1496-1509. [DOD] [CrossRef]
- Muckova K, Duffield JS, Held KD, Bonventre JV, Sheridan AM. cPLA2-interacting protein, PLIP, causes apoptosis and decreases G1 phase in mesangial cells. Am J Physiol Renal Physiol 2006. 290:F70-F79. [DOD] [CrossRef]
|


)
)
)
)
)
)

