Review
| Rev Diabet Stud,
2006,
3(3):108-117 |
DOI 10.1900/RDS.2006.3.108 |
Causes and Characteristics of Diabetic Cardiomyopathy
Jianxun Wang1, Ye Song2, Qianwen Wang3, Patricia M. Kralik2, Paul N. Epstein2
1Departments of Pharmacology and Toxicology, University of Louisville, Louisville, KY 40202, USA.
2Department of Pediatrics, University of Louisville, Louisville, KY 40202, USA.
3Department of Physiology and Biophysics, University of Louisville, Louisville, KY 40202, USA.
Address correspondence to: Paul N. Epstein, e-mail: paul.epstein@louisville.edu
Keywords: diabetes, cardiomyopathy, coronary artery disease, obesity, hyperglycemia, insulin, angiotensin
Abstract
Type 1 and type 2 diabetic patients are at increased risk of cardiomyopathy and heart failure is a major cause of death for these patients. Cardiomyopathy in diabetes is associated with a cluster of features including decreased diastolic compliance, interstitial fibrosis and myocyte hypertrophy. The mechanisms leading to diabetic cardiomyopathy remain uncertain. Diabetes is associated with most known risk factors for cardiac failure seen in the overall population, including obesity, dyslipidemia, thrombosis, infarction, hypertension, activation of multiple hormone and cytokine systems, autonomic neuropathy, endothelial dysfunction and coronary artery disease. In light of these common contributing pathologies it remains uncertain whether diabetic cardiomyopathy is a distinct disease. It is also uncertain which factors are most important to the overall incidence of heart failure in diabetic patients. This review focuses on factors that can have direct effects on diabetic cardiomyocytes: hyperglycemia, altered fuel use, and changes in the activity of insulin and angiotensin. Particular attention is given to the changes these factors can have on cardiac mitochondria and the role of reactive oxygen species in mediating injury to cardiomyocytes.
Recognition of diabetic cardiomyopathy
An association between diabetes and cardiac disease was first recognized in the late 1800s [1]. More recently, the Framingham Heart Study [2] provided conclusive evidence of the role of diabetes in heart failure: in a prospective study of 5,000 individuals, the risk of heart failure was increased in diabetic men and women by two- and five-fold respectively and heart failure is now recognized as a major cause of death among diabetics [3].
Evidence has accumulated indicating that diabetic cardiomyopathy occurs from causes in addition to the coronary atherosclerosis common in diabetes. Other features of diabetes must contribute to the injury to the heart muscle in these patients. This was first proposed by Rubler et al. in 1972 [4] based on postmortem findings of heart failure in diabetic patients free of detectable coronary artery disease. These and similar findings have been reported in many other clinical studies [5, 6]. In addition, the increased risk of diabetic heart failure reported in the Framingham Heart Study could not be fully explained by looking at several other diabetes associated risk factors. Heart studies of general diabetic populations without cardiac complaints [7] also indicated that diabetes produces left ventricular abnormalities independent of other risk factors.
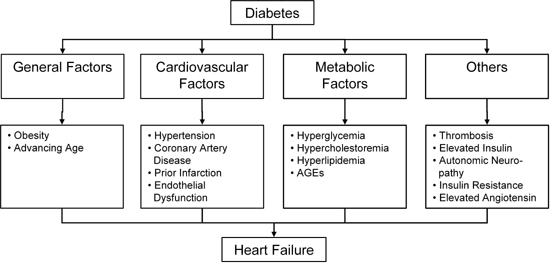
These clinical findings plus many animal studies cited below, suggest that diabetes produces damage to cardiac muscle. Diabetes is associated with most known risk factors for cardiac failure, including obesity, hyperlipidemia, hypercholestoremia, thrombosis, infarction, hypertension, activation of multiple hormone and cytokine systems, autonomic neuropathy, endothelial dysfunction and coronary artery disease (Figure 1). In light of these many contributing pathologies it remains uncertain which factors are most important to the overall incidence of heart failure in diabetic patients.
 |
 |
Figure 1. Some of the risk factors for heart failure which are associated with diabetes. AGE: advanced glycation end product. |
|
Characteristics of diabetic hearts
Cardiomyopathy in type 1 or type 2 diabetic patients is associated with a cluster of common features (Figure 2). It should be noted that these are not unique to diabetes. The most frequent and earliest functional abnormality observed by echocardiography of type 2 diabetic hearts is decreased diastolic compliance [8]. Reduced compliance sometimes co-exists with systolic dysfunction [6], which may be evident as reduced ejection fraction. Diabetes is also a significant risk factor for left ventricular hypertrophy [9] and this has been demonstrated in two large studies of type 2 diabetic populations, both of which reported higher LV wall thickness and mass [2, 10] in diabetic hearts. Hypertrophy was found in both sexes in the Strong Heart Study of American Indians [10] but predominantly in females in the largely Caucasian Framingham Heart Study [2]. Importantly, the ventricular hypertrophy and dysfunction were found to be significant even when accounting for associated risk factors such as BMI and hypertension [10].
 |
|
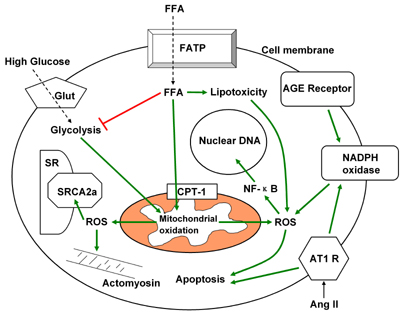
Figure 2. Interacting pathways of ROS production and injury in diabetic cardiomyocytes. ROS: reactive oxygen species. AGE: advanced glycation end products. Ang II: angiotensin II. FFA: free fat acid. Glut: glucose transporter. FATP: free fat acid transport protein. AT1R: angiotensin 2 type 1 receptor. CPT-1: carnitine palmitoyl transferase-1. SR: sarcoplasmic reticulum. SRCA: sarcoplasmic reticulum Ca 2+-ATPase. |
|
Common finding in biopsies of the diabetic heart [11, 12] are interstitial fibrosis and myocyte hypertrophy. Biopsies have also demonstrated diabetes associated increases in contractile protein glycosylation [13]. The biopsy findings of interstitial fibrosis, protein glycosylation and myocyte hypertrophy are likely factors contributing to reduced diastolic compliance and ventricular hypertrophy in diabetic patients. These early signs of cardiac dysfunction, combined with the continuation or exacerbation of other risk factors associated with diabetes, will accelerate the prolonged decline in cardiac function.
In type 1 diabetes, most, though not all [14] studies report diastolic dysfunction. Even young patients in which cardiac ischemia or hypertension could be ruled out, were found to have diastolic dysfunction [15, 16]. In some studies, young type 1 females were more effected than males [16] and pregnancy exacerbates cardiac dysfunction [17]. As seen in most other diabetic complications, there appears to be a correlation between blood glucose control and diastolic dysfunction [18].
Risk factors for diabetic cardiomyopathy
The major reason that diabetics have such a high rate of heart failure can be attributed to their very high rates of the same risk factors that impact the overall population. The most common risk factors for CHF (congestive heart failure) are dyslipidemia and hypertension, both of which are much more frequent in the diabetic population. Prior infarction and re-infarction, while less common than hypertension and dyslipidemia, carry a greater individual risk for development of heart failure. The rate of infarction is several-fold higher in the diabetic population, in part due to their high rates of thrombosis. Coronary atherosclerosis is classically increased in diabetics and many patients have underlying ischemic disease that contributes to both the onset and progression of heart failure. Each of these factors interact, increasing the incidence of CHF more so than any one factor.
Diabetes is also associated with its own set of complications that exacerbate the tendency to cardiomyopathy, including autonomic neuropathy, endothelial dysfunction, peripheral insulin resistance, hyperglycemia and abnormal cardiac fuel usage. Though their individual risk may be low, they are likely to interact with other cardiac pathologies. This review will focus on factors that may have the most direct affect on cardiomyocytes, altered fuel use, hyperglycemia and changes in the activity of insulin and angiotensin.
Mechanisms of diabetic cardiomyopathy
Altered fuel use in diabetic hearts
Healthy hearts derive most of their energy from free fatty acids and only a small proportion from circulating glucose. Typically after an infarction or during heart failure there is an increase in cardiac glucose usage and a reduction in fat consumption. In contrast, diabetic hearts use more fat and less glucose than normal hearts, the opposite of how failing or infarcted hearts modify their fuel use. In one recent study [19] of type 1 diabetic subjects fatty acid uptake was three-fold higher than normal, which coincided with a two-fold decrease in glucose uptake. Similar results have been demonstrated in several rodent models of type 2 diabetes [20]. The cardiac metabolic switch for increased fat consumption appears to be related to high circulating levels of fatty acids and reduced cardiac glucose usage. The whole body shift to reduced glucose usage and increased fatty acid availability occurs in diabetes because of reduced insulin action in several tissues. Increased release of fatty acids by the adipocyte and liver results in elevated circulating fatty acids and triglycerides which is a major factor in the elevated uptake and oxidation of fatty acids in cardiomyocytes [21]. To adapt to the requirement for increased fatty acid oxidation, hearts from type 1 diabetic models show dramatic up-regulation of mitochondrial proteins involved in fat metabolism [22, 23]. The induction of enzymes of fatty acid oxidation requires the transcriptional regulators PPARα and/or PPARβ, which regulate gene expression of these enzymes [24]. These PPAR factors are elevated and activated in diabetic hearts [25].
Increased fat dependence also appears in part to be a function of decreased glucose metabolism. The presence of glucose reduces fatty acid metabolism, probably by increasing intracellular levels of malonyl CoA [26], a potent inhibitor of fatty acid conjugation to carnitine. This step is the major control point for movement of fatty acids into the mitochondria for oxidation. Cardiac glucose metabolism declines in diabetes [19] due to a decline in insulin, insulin resistance or increased availability of fatty acids. The first step in cardiomyocyte glucose usage is uptake which is significantly regulated by insulin in the heart. In diabetes there is also a chronic reduction in cardiac glycolytic capacity [27] and glucose oxidation is still further reduced by a decline in pyruvate dehydrogenase activity [27, 28].
Bishop and Altrud reported over 30 years ago [29] that glycolytic metabolism is increased in cardiac hypertrophy and congestive heart failure. This may be an adaptive response that allows for increased energy efficiency, as well more ATP production by way of anerobic glycolysis. The significance of this increased requirement of glucose during heart failure has been indicated in several systems where glucose metabolism has been modified. Impaired glycolysis by transgenic inhibition of phosphofructokinase [30] predisposes to heart failure. The most prominent finding in glucose transporter (GLUT4) knockout mice is cardiac hypertrophy [31]. Apoptosis of cardiac myocytes, which is known to occur during heart failure [32], is inhibited following modifications that increase glycolysis [33]. Studies utilizing in vitro application of inhibitors of glucose metabolism, such as 2-deoxyglucose [34] report weakened contractility, even under well-oxygenated conditions. Notably, the most prominent finding described following inhibition of glycolysis was impairment of diastolic relaxation, which is also the most prominent defect in diabetic cardiomyopathy. Conversely, transgenic manipulations that increase glucose usage provides protection from heart failure due to aortic constriction [35] or diabetes [36].
Reduced glycolytic activity of the diabetic heart may also be an important factor that predisposes diabetic patients to more severe outcomes following ischemic or hypoxic damage. It was shown 30 years ago in ischemic rat hearts [37], that dependence on glucose and glycolysis is markedly increased. One of the protective responses of the ischemic heart is an increase in glycolysis [38, 39] and stimulation of glycolysis with glucose and insulin has been used for many decades to protect patient hearts from ischemic or hypoxic damage [40, 41]. In vitro experiments to examine the relationship between glycolytic rate and ischemic injury support this clinical practice. Studies in isolated perfused hearts reveal that manipulations which accelerate cardiac glucose use decrease ischemic damage [42, 43] while procedures that limit glycolysis tend to sensitize the heart to ischemia [44]. Tian et al. [45] found that cardiac specific knockout of the GLUT4 glucose transporter, sensitizes the heart to hypoxic damage. The mechanism of glucose-induced cardiac protection in ischemia has not been resolved. However, it is clear that glycolysis becomes the sole or primary source of ATP production in hypoxic hearts. Also, ATP derived from glycolysis seems to have a preferential role [46] in maintaining normal conductance for calcium, potassium and sodium ions, functions that are critical in maintaining cardiac myocytes viability during ischemia.
The pathological significance of this increased dependence on fatty acid metabolism in the diabetic cardiomyocyte remains an uncertain but suspect cause for cardiomyopathy. Transgenic studies that produce heart damage by way of cardiac specific increases in fat metabolism, indicate that injury originates in the myocyte and is not simply secondary to systemic changes [47] associated with hyperlipidemia. Excessive dependence on fatty acid metabolism poses several problems for the heart. Increased intracellular fatty acids can be detrimental to mitochondria [48]. The fatty acid palmitate, possibly by conversion to ceramide [49] is particularly potent in inducing apoptosis in cardiomyocytes [50] and palmitate exposure also damages the contractile apparatus [49]. In addition, excessive reliance on fatty acid metabolism impairs cardiac energy efficiency at least in part because glucose utilization is about 10% more efficient at generating ATP per O2 consumed (2.58 vs. 2.33 ATP/oxygen atom). This change in efficiency impacts the heart of diabetic and obese patients. Obesity in young women has been shown to be correlated with increased fatty acid utilization, increased cardiac oxygen consumption, and decreased cardiac efficiency [51]. In type 1 diabetic hearts there is also an increased consumption of oxygen [19].
Studies on mitochondria from hearts of diabetic animal models demonstrate morphological and functional changes that may be secondary to the shift in greater fat availability. Diabetes alters the protein composition of diabetic mitochondria [52] to accommodate the increased oxidation of fatty acids. Proteomic analysis of cardiac proteins altered by diabetes revealed that 60% of the proteins that increased in abundance were localized to mitochondria, which is a striking finding considering that only 1-2% of cellular proteins are mitochondrial [52]. Most of the protein changes were due to increased content of enzymes required for fatty acid oxidation. It has also been proposed that the increase in fat dependence may also disturb mitochondrial lipid composition [53]. Consistent with those findings, diabetes also reduces mitochondrial efficiency for ATP production [52]. Diabetic mitochondria also produce more ROS than normal [54] which can further damage mitochondria thereby decreasing efficiency. In support of this, it was found that transgenic over-expression of the mitochondrial antioxidant enzyme manganese superoxide dismutase partially restored normal function in mitochondria from diabetic hearts [55].
Intracellular lipid accumulation
If fatty acid oxidation fails to keep up with uptake, lipids can accumulate, producing lipotoxicity [56, 57]. The Taegtmeyer group [58] has observed a significant accumulation of lipid in cardiac myocytes of heart failure patients. Not surprisingly, this was most evident in diabetic patients, to a lesser extent in obese patients and not at all in non-obese, non-diabetic patients. This suggests that lipid accumulation does not play a role in many cases of heart failure but may be an important factor in patients that are obese or diabetic. Lipid accumulation may directly impede myocyte metabolism and contractility or promote cardiomyocyte apoptosis.
A combination of increased cardiomyocyte fatty acid transport proteins [57], increased lipoprotein lipase [47] or elevated serum fatty acids will facilitate the import of fatty acids or neutral lipids into the cardiomyocyte. There is an adaptive process for this fuel surplus, likely by lipid ligand activation of PPAR alpha and beta with subsequent gene expression, particularly those involved in fatty acid oxidation [24]. However, the implication that more PPAR alpha activity, as seen in diabetic hearts [25], will be beneficial does not seem to be true. For example, Taegetmeyer [59] has recently shown that PPAR alpha activation exacerbates lipotoxicity in a mouse model of repetitive ischemia reperfusion. Also, cardiac specific over-expression of PPAR alpha receptors [60] exacerbates diabetes-induced cardiomyopathy, while whole body knockout of PPAR alpha limited induction of diabetic cardiomyopathy. These results emphasize that cardiac lipid balance is carefully regulated and is readily disturbed, as occurs in diabetes.
The mechanism of damage due to lipid accumulation is not certain; however Unger has emphasized the concept that only adipocytes are competent for extensive storage of lipids whereas all other cell types are susceptible to lipotoxic injury [61]. Accumulation of palmitate in cardiomyoblasts produces both increased ROS production and ER stress, resulting in apoptosis [62]. Palmitate accumulation can also promote denovo ceramide production [63] which is also an inducer of apoptosis. Fatty acids can also modify intracellular signaling mechanisms. For example, in skeletal muscle, free fatty acids promote insulin resistance by altering signaling at multiple steps in the insulin activation cascade [63]. These apparently different mechanisms of cell injury may be independent mechanisms of damage or they may be interdependent and sequential.
Altered cell signaling in diabetic hearts
Organ sensitivity to hormones and the hormonal milieu are markedly altered in diabetes. Circulating insulin levels are very high, especially early in type 2 diabetes, to compensate for insulin resistance in skeletal muscle, adipocytes and the liver. While it is by no means established, it appears that cardiac cells of diabetic patients do not develop insulin resistance to the same extent as seen in skeletal muscle. Some studies have not observed insulin resistance in hearts of type 2 diabetic patients, even when other diabetic tissues of the same patients exhibit significant insulin resistance [64, 65]. Thus under conditions of diabetic hyperinsulinemia, the less-resistant heart is being stimulated by relatively high levels of insulin. Insulin is known to promote the actions of various growth factors and increase in cell growth in multiple cell types [66]. In cardiomyocytes, insulin stimulates hypertrophy by several pathways, notably activation of Akt and ERK. This can be expected to be one factor in the hypertrophy characteristic of diabetic hearts. Over stimulation of some insulin responses may also be true in type 1 diabetics if the dose of insulin is titrated to tissue that are more insulin resistant that the heart.
Angiotensin 2 has been implicated in several complications of diabetes and blockade of angiotensin synthesis or its receptor is recognized as the most effective preventative treatment for diabetic nephropathy. The Anversa group [67] has demonstrated increased angiotensin 2 labeling of cardiac myocytes and endothelial cells in cardiac biopsy samples of patients with type 2 diabetes. They have also shown increased expression of synthetic and receptor components of the rennin-angiotensin system in an animal model of type 1 diabetes [68]. Angiotenin 2 increases ROS production in cardiomyocytes and we have found that type 1 diabetic mouse cardiomyocytes produce more ROS than normal cardiomyocytes [54]. Angiotensin 2 is also capable of inducing two of the most characteristic features of diabetic cardiomyopathy, hypertrophy and interstitial fibrosis [69]. Fibrosis may in part be due to activation of connective tissue growth factor [69] and TGF-β activity, as well as by induction of plasminogen activator inhibitor-1, which attenuates fibrinolysis. Angiotensin stimulation of ROS production may occur via local activation of NADPH oxidase [70] or by increased mitochondrial ROS generation [54]. Excess ROS that produces cardiomyocyte death will further induce replacement fibrosis.
High glucose-induced generation of AGEs and reactive oxygen species in diabetic hearts
Elevated circulating glucose has been shown to be a major factor in most complications of type 1 [71] and type 2 diabetes [72]. Those clinical trials did not analyze diabetic cardiomyopathy but extensive evidence from experimental models of type 1 and 2 diabetes implicates hyperglycemia in cardiomyopathy. Important components of high glucose-induced cellular injury are the generation of reactive oxygen species (ROS) and formation of advanced glycation end products (AGEs). AGEs are formed when glucose or glucose metabolites produce stable, covalent modification of proteins. These protein adducts are not only directly damaging but they also contribute to ROS generation.
In the extra-cellular compartment, the major AGEs derive from direct reaction of glucose with protein amino groups. The modifications can be a single, isolated change on the peptide chain or multiple AGE modifications that can produce crosslinks within or between proteins. Long-lived extra-cellular proteins such as collagen and elastin are particularly vulnerable to accumulation of AGE crosslinks [73]. This can impair the ability of collagen to be degraded, leading to collagen accumulation or fibrosis. Crosslinks in collagen and elastin and the resulting fibrosis also cause increased myocardial stiffness and impaired cardiac relaxation, typical of diabetic hearts. In a rodent diabetic model, treatment with the crosslink breaker, ALT-711, reduced cardiac AGE levels, improved collagen solubility and ameliorated diabetes-induced changes in cardiac gene expression [74]. While not yet studied specifically in human diabetic patients, it has been shown in patients with diastolic heart failure, which is characteristic of diabetic cardiomyopathy, that the crosslink breaker, ALT-711 improves diastolic performance and reduces cardiac hypertrophy [75]. Thus, crosslink breakers provide what is theoretically a very promising class of drugs for treatment and prevention of diabetic cardiomyopathy.
Soluble extra-cellular AGEs can bind to several different cell surface receptors. The most important of these with respect to diabetic complications is the receptor for advanced glycation end products, RAGE [76]. This receptor binds to a broad class of ligands including AGEs and the resulting RAGE activation stimulates NADPH oxidase and other intracellular signaling pathways [76]. Activated NADPH oxidase produces large amounts of cytoplasmic and extra-cellular superoxide which in turn can combine with nitric oxide, forming highly reactive and damaging peroxynitrite. Since nitric oxide is an important cell signaling molecule, the reduction in nitric oxide levels due to formation of peroxynitrite impairs normal cell signaling. Superoxide also converts to another highly reactive ROS, the hydroxyl radical which can damage proteins, lipids and nucleic acids. By elevation of intracellular free radicals and by the triggering of multiple other signaling pathways [76], the activated RAGE receptor up-regulates the stress related transcription factor NF-κB [77] and modifies overall cellular gene expression. RAGE receptors are found in cardiomyocytes [78] and they are a significant component of cardiac ischemia reperfusion injury in rodent models [79]. In addition, transgenic over-expression of RAGE in the heart produces features of rodent models of diabetic cardiomyopathy such as reduced intracellular calcium transients and prolongation of calcium peaks [78]. The RAGE receptor provides another target for potential therapeutics for diabetic complications [80] including cardiomyopathy.
Accumulated evidence from many laboratories, particularly the Brownlee laboratory [81] indicate that another important contributor to cell injury is the increased level of glucose that enters the cell in diabetes and goes through mitochondrial oxidation. The resultant increase in mitochondrial metabolism causes more production of superoxide from the mitochondrial electron transport chain. Mitochondrial generated superoxide, like superoxide produced by NADPH oxidase, generates highly reactive free radicals that damage DNA, protein and lipid constituents of the cell. Much of the evidence for this concept of diabetes-induced damage derives from endothelial cells. This is a very appropriate cell type, since it is basic to the impaired function of blood vessels and microcirculation in complications such as nephropathy and retinopathy, which are responsive to levels of glycemia. Endothelial cells utilize glucose transporters that are largely insulin independent; their glucose uptake is more affected by the glucose gradient across the cytoplasmic membrane and less effected by ambient insulin or by insulin resistance. Thus in endothelial cells, elevations in extra-cellular glucose produce increases in intracellular glucose.
It is less certain whether diabetes induces the same elevation in intracellular glucose in cardiac cells as it does in endothelial cells. Unlike endothelial cells, skeletal and cardiac muscle utilize the insulin responsive GLUT 4 transporter. Thus when insulin levels or insulin responsiveness is low, the capacity for glucose uptake in muscle is reduced and intracellular glucose levels are less likely to rise dramatically. However, the heart has minimal or at least a lower degree of insulin resistance than skeletal muscle in type 2 diabetes [64, 65]. As a result, cardiac glucose transporters may not be down-regulated in diabetic patients. There are discrepant findings with regard to glucose uptake in hearts of diabetic patients; some studies report decreased glucose uptake [82] while other studies in type 2 diabetic subjects report normal glucose uptake [64]. If glucose transport is normal and extra-cellular glucose is elevated, then it may be expected that intracellular glucose will rise above normal in cardiomyocytes of type 2 diabetic patients. AGE formation is much more rapid with intermediate metabolites of glycolysis than it is with glucose itself. However, it has not been established whether the concentration of individual reactive metabolites is altered in hearts of diabetic patients. One indicator of increased cardiac intracellular glucose or its metabolites would be to determine a concurrent increase in intracellular AGE-modified cardiomyocyte proteins. This has been definitively demonstrated, at least in diabetic rats [83]. Cardiac sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA) was found to contain an increase in single and crosslinked AGEs. This has the potential to impair the capacity of SERCA to translocate calcium and thus slow the rate of cardiac relaxation. Both delayed calcium uptake and relaxation are characteristic features of diabetic hearts [84]. In addition, our laboratory [54] and others [70] have found that cardiomyocytes from diabetic mice exposed to high glucose, produce more ROS than normal cardiomyocytes generated by mitochondrial metabolism. These findings of high glucose-induced ROS production and intracellular AGE content suggest that cardiomyocytes are sensitive to intracellular glucose toxicity in diabetes. Figure 2 summarizes the major sources and targets of ROS in diabetic cardiomyocytes.
Conclusions
Basic questions remain in understanding diabetic cardiomyopathy, including determining whether it is distinct from other forms of heart failure. The main clinical findings of hypertrophy and impaired diastolic function are by no means unique. Only with a pre-existing diagnosis of diabetes can diabetic cardiomyopathy be diagnosed. This leads to an ongoing disagreement among clinicians and basic scientists whether diabetic cardiomyopathy is a distinct pathology. Despite the fact that we do not yet have a unique description for a constellation of pathologies defining diabetic cardiomyopathy, it is clearly a very serious clinical problem simply based on the two- to five-fold increased rates of heart failure among diabetics. Diabetics have high rates of all the common risk factors for heart failure (Table 1). In addition, diabetics have two risk factors that are related to one another but distinct from the non-diabetic population, hyperglycemia and high levels of AGEs. Hyperglycemia accounts for part of the risk of diabetic heart failure: for each 1% increase in glycosylated hemoglobin in the range of 7 to 10% there is approximately an 8% increase in the risk for heart failure [85]. This is significant, but does not account for the 100% to 300% risk added by diabetes [2]. It is likely that hyperglycemina combines with the more common features to multiply the overall risk. As so many of these risk factors are associated with each other it is a daunting task even for the largest and most well designed multivariate analysis to determine the relative importance of each individual factor. It also remains to be determined why diabetes has a more than two-fold greater effect on heart failure in women than in men [86]. Answers to these questions will most likely come first from basic research in animal models. Many different diabetic models are available but they display significantly different cardiac phenotypes [63]. The sort of focused phenotyping centers such as those supported by the NIH identifying optimal models of diabetic nephropathy and retinopathy will be valuable in identifying an optimal model displaying similar features seen in human diabetics. This will produce more consistent and meaningful findings on the causes of diabetic cardiomyopathy.
Table
1.
Clinical features of diabetic cardiomyopathy |
|
 |
|
Acknowledgments:
Supported by NIH grants DK073586 and HL62892.
References
- Vergely P. De l'angine de poitrine dans ses rapports avec le diabete. Gaz Hebd Med Chir 1883. 20:364-368. [DOD]
- Kannel WB, Hjortland M, Castelli WP. Role of diabetes in congestive heart failure: the Framingham study. Am J Cardiol 1974. 34(1):29-34. [DOD] [CrossRef]
- Abbott RD, Donahue RP, Kannel WB, Wilson PW. The impact of diabetes on survival following myocardial infarction in men vs. women. The Framingham Study. JAMA 1988. 260(23):3456-3460. [DOD] [CrossRef]
- Rubler S, Dlugash J, Yuceoglu YZ, Kumral T, Branwood AW, Grishman A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am J Cardiol 1972. 30(6):595-602. [DOD] [CrossRef]
- Hamby RI, Zoneraich S, Sherman L. Diabetic cardiomyopathy. JAMA 1974. 229(13):1749-1754. [DOD] [CrossRef]
- Regan TJ, Lyons MM, Ahmed SS, Levinson GE, Oldewurtel HA, Ahmad MR, Haider B. Evidence for cardiomyopathy in familial diabetes mellitus. J Clin Invest 1977. 60(4):884-899. [DOD]
- Bell DS. Diabetic cardiomyopathy. A unique entity or a complication of coronary artery disease? Diabetes Care 1995. 18(5):708-714. [DOD]
- Shehadeh A, Regan TJ. Cardiac consequences of diabetes mellitus. Clin Cardiol 1995. 18(6):301-305. [DOD]
- Galderisi M, Anderson KM, Wilson PW, Levy D. Echocardiographic evidence for the existence of a distinct diabetic cardiomyopathy (the Framingham Heart Study). Am J Cardiol 1991. 68(1):85-89. [DOD] [CrossRef]
- Devereux RB, Roman MJ, Paranicas M, O'Grady MJ, Lee ET, Welty TK, Fabsitz RR, Robbins D, Rhoades ER, Howard BV. Impact of diabetes on cardiac structure and function: the strong heart study. Circulation 2000. 101(19):2271-2276. [DOD]
- Nunoda S, Genda A, Sugihara N, Nakayama A, Mizuno S, Takeda R. Quantitative approach to the histopathology of the biopsied right ventricular myocardium in patients with diabetes mellitus. Heart Vessels 1985. 1(1):43-47. [DOD] [CrossRef]
- Das AK, Das JP, Chandrasekar S. Specific heart muscle disease in diabetes mellitus - a functional structural correlation. Int J Cardiol 1987. 17(3):299-302. [DOD] [CrossRef]
- Syrovy I, Hodny Z. Non-enzymatic glycosylation of myosin: effects of diabetes and ageing. Gen Physiol Biophys 1992. 11(3):301-307. [DOD]
- Romanens M, Fankhauser S, Saner B, Michaud L, Saner H. No evidence for systolic or diastolic left ventricular dysfunction at rest in selected patients with long-term type I diabetes mellitus. Eur J Heart Fail 1999. 1(2):169-175. [DOD] [CrossRef]
- Schannwell CM, Schneppenheim M, Perings S, Plehn G, Strauer BE. Left ventricular diastolic dysfunction as an early manifestation of diabetic cardiomyopathy. Cardiology 2002. 98(1-2):33-39. [DOD] [CrossRef]
- Suys BE, Katier N, Rooman RP, Matthys D, Op De BL, Du Caju MV, De WD. Female children and adolescents with type 1 diabetes have more pronounced early echocardiographic signs of diabetic cardiomyopathy. Diabetes Care 2004. 27(8):1947-1953. [DOD]
- Schannwell CM, Schneppenheim M, Perings SM, Zimmermann T, Plehn G, Strauer BE. Alterations of left ventricular function in women with insulin-dependent diabetes mellitus during pregnancy. Diabetologia 2003. 46(2):267-275. [DOD]
- Shishehbor MH, Hoogwerf BJ, Schoenhagen P, Marso SP, Sun JP, Li J, Klein AL, Thomas JD, Garcia MJ. Relation of hemoglobin A1c to left ventricular relaxation in patients with type 1 diabetes mellitus and without overt heart disease. Am J Cardiol 2003. 91(12):1514-1517. [DOD] [CrossRef]
- Herrero P, Peterson LR, McGill JB, Matthew S, Lesniak D, Dence C, Gropler RJ. Increased myocardial fatty acid metabolism in patients with type 1 diabetes mellitus. J Am Coll Cardiol 2006. 47(3):598-604. [DOD] [CrossRef]
- Carley AN, Severson DL. Fatty acid metabolism is enhanced in type 2 diabetic hearts. Biochim Biophys Acta 2005. 1734(2):112-126. [DOD]
- Young ME, Guthrie PH, Razeghi P, Leighton B, Abbasi S, Patil S, Youker KA, Taegtmeyer H. Impaired long-chain fatty acid oxidation and contractile dysfunction in the obese Zucker rat heart. Diabetes 2002. 51(8):2587-2595. [DOD]
- Shen X, Ye G, Metreveli NS, Epstein PN. Cardiomyocyte defects in diabetic models and protection with cardiac-targeted transgenes. Methods Mol Med 2005. 112:379-388. [DOD]
- Turko IV, Murad F. Quantitative protein profiling in heart mitochondria from diabetic rats. J Biol Chem 2003. 278(37):35844-35849. [DOD] [CrossRef]
- Huss JM, Kelly DP. Mitochondrial energy metabolism in heart failure: a question of balance. J Clin Invest 2005. 115(3):547-555. [DOD] [CrossRef]
- Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, Han X, Gross RW, Kozak R, Lopaschuk GD, Kelly DP. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J Clin Invest 2002. 109(1):121-130. [DOD] [CrossRef]
- Awan MM, Saggerson ED. Malonyl-CoA metabolism in cardiac myocytes and its relevance to the control of fatty acid oxidation. Biochem J 1993. 295(Pt 1):61-66. [DOD]
- Stanley WC, Lopaschuk GD, McCormack JG. Regulation of energy substrate metabolism in the diabetic heart. Cardiovasc Res 1997. 34(1):25-33. [DOD] [CrossRef]
- Huang B, Wu P, Popov KM, Harris RA. Starvation and diabetes reduce the amount of pyruvate dehydrogenase phosphatase in rat heart and kidney. Diabetes 2003. 52(6):1371-1376. [DOD]
- Bishop SP, Altschuld RA. Increased glycolytic metabolism in cardiac hypertrophy and congestive failure. Am J Physiol 1970. 218(1):153-159. [DOD]
- Donthi RV, Ye G, Wu C, McClain DA, Lange AJ, Epstein PN. Cardiac expression of kinase-deficient 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase inhibits glycolysis, promotes hypertrophy, impairs myocyte function, and reduces insulin sensitivity. J Biol Chem 2004. 279(46):48085-48090. [DOD] [CrossRef]
- Stenbit AE, Tsao TS, Li J, Burcelin R, Geenen DL, Factor SM, Houseknecht K, Katz EB, Charron MJ. GLUT4 heterozygous knockout mice develop muscle insulin resistance and diabetes. Nat Med 1997. 3(10):1096-1101. [DOD] [CrossRef]
- Narula J, Haider N, Virmani R, DiSalvo TG, Kolodgie FD, Hajjar RJ, Schmidt U, Semigran MJ, Dec GW, Khaw BA. Apoptosis in myocytes in end-stage heart failure. N Engl J Med 1996. 335(16):1182-1189. [DOD] [CrossRef]
- Malhotra R, Brosius FC 3rd. Glucose uptake and glycolysis reduce hypoxia-induced apoptosis in cultured neonatal rat cardiac myocytes. J Biol Chem 1999. 274(18):12567-12575. [DOD] [CrossRef]
- Kagaya Y, Weinberg EO, Ito N, Mochizuki T, Barry WH, Lorell BH. Glycolytic inhibition: effects on diastolic relaxation and intracellular calcium handling in hypertrophied rat ventricular myocytes. J Clin Invest 1995. 95(6):2766-2776. [DOD]
- Liao R, Jain M, Cui L, D'Agostino J, Aiello F, Luptak I, Ngoy S, Mortensen RM, Tian R. Cardiac-specific overexpression of GLUT1 prevents the development of heart failure attributable to pressure overload in mice. Circulation 2002. 106(16):2125-2131. [DOD] [CrossRef]
- Belke DD, Larsen TS, Gibbs EM, Severson DL. Altered metabolism causes cardiac dysfunction in perfused hearts from diabetic (db/db) mice. Am J Physiol Endocrinol Metab 2000. 279(5):E1104-E1113. [DOD]
- Neely JR, Rovetto MJ. Techniques for perfusing isolated rat hearts. Methods Enzymol 1975. 39:43-60. [DOD]
- Russell RR 3rd, Yin R, Caplan MJ, Hu X, Ren J, Shulman GI, Sinusas AJ, Young LH. Additive effects of hyperinsulinemia and ischemia on myocardial GLUT1 and GLUT4 translocation in vivo. Circulation 1998. 98(20):2180-2186. [DOD]
- Stanley WC, Lopaschuk GD, Hall JL, McCormack JG. Regulation of myocardial carbohydrate metabolism under normal and ischaemic conditions. Potential for pharmacological interventions. Cardiovasc Res 1997. 33(2):243-257. [DOD] [CrossRef]
- Oldfield GS, Commerford PJ, Opie LH. Effects of preoperative glucose-insulin-potassium on myocardial glycogen levels and on complications of mitral valve replacement. J Thorac Cardiovasc Surg 1986. 91(6):874-878. [DOD]
- Opie LH. Glucose and the metabolism of ischaemic myocardium. Lancet 1995. 345(8964):1520-1521. [DOD] [CrossRef]
- Owen P, Dennis S, Opie LH. Glucose flux rate regulates onset of ischemic contracture in globally underperfused rat hearts. Circ Res 1990. 66(2):344-354. [DOD]
- Vanoverschelde JL, Janier MF, Bakke JE, Marshall DR, Bergmann SR. Rate of glycolysis during ischemia determines extent of ischemic injury and functional recovery after reperfusion. Am J Physiol 1994. 267(5 Pt 2):H1785-H1794. [DOD]
- Jeremy RW, Ambrosio G, Pike MM, Jacobus WE, Becker LC. The functional recovery of post-ischemic myocardium requires glycolysis during early reperfusion. J Mol Cell Cardiol 1993. 25(3):261-276. [DOD] [CrossRef]
- Tian R, Pham M, Abel ED. Cardiac glucose transporter deficiency increases the susceptibility of the heart to ischemic injury. Diabetes 1999. Abstracts of 59th Scientific Session 1999. A127. [DOD]
- Weiss J, Hiltbrand B. Functional compartmentation of glycolytic versus oxidative metabolism in isolated rabbit heart. J Clin Invest 1985. 75(2):436-447. [DOD]
- Yagyu H, Chen G, Yokoyama M, Hirata K, Augustus A, Kako Y, Seo T, Hu Y, Lutz EP, Merkel M, Bensadoun A, Homma S, Goldberg IJ. Lipoprotein lipase (LpL) on the surface of cardiomyocytes increases lipid uptake and produces a cardiomyopathy. J Clin Invest 2003. 111(3):419-426. [DOD] [CrossRef]
- Di Paola M, Lorusso M. Interaction of free fatty acids with mitochondria: Coupling, uncoupling and permeability transition. Biochim Biophys Acta 2006. 1757(9-10):1330-1337. [DOD] [CrossRef]
- Dyntar D, Eppenberger-Eberhardt M, Maedler K, Pruschy M, Eppenberger HM, Spinas GA, Donath MY. Glucose and palmitic acid induce degeneration of myofibrils and modulate apoptosis in rat adult cardiomyocytes. Diabetes 2001. 50(9):2105-2113. [DOD]
- Sparagna GC, Jones CE, Hickson-Bick DL. Attenuation of fatty acid-induced apoptosis by low-dose alcohol in neonatal rat cardiomyocytes. Am J Physiol Heart Circ Physiol 2004. 287(5):H2209-H2215. [DOD] [CrossRef]
- Peterson LR, Herrero P, Schechtman KB, Racette SB, Waggoner AD, Kisrieva-Ware Z, Dence C, Klein S, Marsala J, Meyer T, Gropler RJ. Effect of obesity and insulin resistance on myocardial substrate metabolism and efficiency in young women. Circulation 2004. 109(18):2191-2196. [DOD] [CrossRef]
- Shen X, Zheng S, Thongboonkerd V, Xu M, Pierce WM Jr, Klein JB, Epstein PN. Cardiac mitochondrial damage and biogenesis in a chronic model of type 1 diabetes. Am J Physiol Endocrinol Metab 2004. 287(5):E896-E905. [DOD] [CrossRef]
- Su X, Han X, Mancuso DJ, Abendschein DR, Gross RW. Accumulation of long-chain acylcarnitine and 3-hydroxy acylcarnitine molecular species in diabetic myocardium: identification of alterations in mitochondrial fatty acid processing in diabetic myocardium by shotgun lipidomics. Biochemistry 2005. 44(13):5234-5245. [DOD] [CrossRef]
- Ye G, Metreveli NS, Donthi RV, Xia S, Xu M, Carlson EC, Epstein PN. Catalase protects cardiomyocyte function in models of type 1 and type 2 diabetes. Diabetes 2004. 53(5):1336-1343. [DOD]
- Shen X, Zheng S, Metreveli NS, Epstein PN. Protection of cardiac mitochondria by overexpression of MnSOD reduces diabetic cardiomyopathy. Diabetes 2006. 55(3):798-805. [DOD] [CrossRef]
- Cheng L, Ding G, Qin Q, Huang Y, Lewis W, He N, Evans RM, Schneider MD, Brako FA, Xiao Y, Chen YE, Yang Q. Cardiomyocyte-restricted peroxisome proliferator-activated receptor-delta deletion perturbs myocardial fatty acid oxidation and leads to cardiomyopathy. Nat Med 2004. 10(11):1245-1250. [DOD] [CrossRef]
- Chiu HC, Kovacs A, Blanton RM, Han X, Courtois M, Weinheimer CJ, Yamada KA, Brunet S, Xu H, Nerbonne JM, Welch MJ, Fettig NM, Sharp TL, Sambandam N, Olson KM, Ory DS, Schaffer JE. Transgenic expression of fatty acid transport protein 1 in the heart causes lipotoxic cardiomyopathy. Circ Res 2005. 96(2):225-233. [DOD] [CrossRef]
- Sharma S, Adrogue JV, Golfman L, Uray I, Lemm J, Youker K, Noon GP, Frazier OH, Taegtmeyer H. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J 2004. 18(14):1692-1700. [DOD] [CrossRef]
- Dewald O, Sharma S, Adrogue J, Salazar R, Duerr GD, Crapo JD, Entman ML, Taegtmeyer H. Downregulation of peroxisome proliferator-activated receptor-alpha gene expression in a mouse model of ischemic cardiomyopathy is dependent on reactive oxygen species and prevents lipotoxicity. Circulation 2005. 112(3):407-415. [DOD] [CrossRef]
- Finck BN, Han X, Courtois M, Aimond F, Nerbonne JM, Kovacs A, Gross RW, Kelly DP. A critical role for PPARalpha-mediated lipotoxicity in the pathogenesis of diabetic cardiomyopathy: modulation by dietary fat content. Proc Natl Acad Sci U S A 2003. 100(3):1226-1231. [DOD] [CrossRef]
- Unger RH. Lipotoxic diseases. Annu Rev Med 2002. 53:319-336. [DOD] [CrossRef]
- Borradaile NM, Han X, Harp JD, Gale SE, Ory DS, Schaffer JE. Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J Lipid Res 2006. In press. [DOD]
- Poornima IG, Parikh P, Shannon RP. Diabetic cardiomyopathy: the search for a unifying hypothesis. Circ Res 2006. 98(5):596-605. [DOD] [CrossRef]
- Utriainen T, Takala T, Luotolahti M, Ronnemaa T, Laine H, Ruotsalainen U, Haaparanta M, Nuutila P, Yki-Jarvinen H. Insulin resistance characterizes glucose uptake in skeletal muscle but not in the heart in NIDDM. Diabetologia 1998. 41(5):555-559. [DOD] [CrossRef]
- Jagasia D, Whiting JM, Concato J, Pfau S, McNulty PH. Effect of non-insulin-dependent diabetes mellitus on myocardial insulin responsiveness in patients with ischemic heart disease. Circulation 2001. 103(13):1734-1739. [DOD]
- Hsueh WA, Law RE. Insulin signaling in the arterial wall. Am J Cardiol 1999. 84(1A):21J-24J. [DOD] [CrossRef]
- Frustaci A, Kajstura J, Chimenti C, Jakoniuk I, Leri A, Maseri A, Nadal-Ginard B, Anversa P. Myocardial cell death in human diabetes. Circ Res 2000. 87(12):1123-1132. [DOD]
- Fiordaliso F, Li B, Latini R, Sonnenblick EH, Anversa P, Leri A, Kajstura J. Myocyte death in streptozotocin-induced diabetes in rats in angiotensin II- dependent. Lab Invest 2000. 80(4):513-527. [DOD]
- He Z, Way KJ, Arikawa E, Chou E, Opland DM, Clermont A, Isshiki K, Ma RC, Scott JA, Schoen FJ, Feener EP, King GL. Differential regulation of angiotensin II-induced expression of connective tissue growth factor by protein kinase C isoforms in the myocardium. J Biol Chem 2005. 280(16):15719-15726. [DOD] [CrossRef]
- Kajstura J, Fiordaliso F, Andreoli AM, Li B, Chimenti S, Medow MS, Limana F, Nadal-Ginard B, Leri A, Anversa P. IGF-1 overexpression inhibits the development of diabetic cardiomyopathy and angiotensin II-mediated oxidative stress. Diabetes 2001. 50(6):1414-1424. [DOD]
- The Diabetes Control and Complications Trial Research Group. The Effect of Intensive Treatment of Diabetes on the Development and Progression of Long-Term Complications in Insulin-Dependent Diabetes Mellitus. N Engl J Med 1993. 329(14):977-986. [DOD] [CrossRef]
- UK Prospective Diabetes Study Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998. 352(9131):837-853. [DOD] [CrossRef]
- Susic D, Varagic J, Ahn J, Frohlich ED. Collagen cross-link breakers: a beginning of a new era in the treatment of cardiovascular changes associated with aging, diabetes, and hypertension. Curr Drug Targets Cardiovasc Haematol Disord 2004. 4(1):97-101. [DOD] [CrossRef]
- Candido R, Forbes JM, Thomas MC, Thallas V, Dean RG, Burns WC, Tikellis C, Ritchie RH, Twigg SM, Cooper ME, Burrell LM. A breaker of advanced glycation end products attenuates diabetes-induced myocardial structural changes. Circ Res 2003. 92(7):785-792. [DOD] [CrossRef]
- Little WC, Zile MR, Kitzman DW, Hundley WG, O'Brien TX, Degroof RC. The effect of alagebrium chloride (ALT-711), a novel glucose cross-link breaker, in the treatment of elderly patients with diastolic heart failure. J Card Fail 2005. 11(3):191-195. [DOD] [CrossRef]
- Goldin A, Beckman JA, Schmidt AM, Creager MA. Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation 2006. 114(6):597-605. [DOD] [CrossRef]
- Bierhaus A, Chevion S, Chevion M, Hofmann M, Quehenberger P, Illmer T, Luther T, Berentshtein E, Tritschler H, Muller M, Wahl P, Ziegler R, Nawroth PP. Advanced glycation end product-induced activation of NF-kappaB is suppressed by alpha-lipoic acid in cultured endothelial cells. Diabetes 1997. 46(9):1481-1490. [DOD]
- Petrova R, Yamamoto Y, Muraki K, Yonekura H, Sakurai S, Watanabe T, Li H, Takeuchi M, Makita Z, Kato I, Takasawa S, Okamoto H, Imaizumi Y, Yamamoto H. Advanced glycation endproduct-induced calcium handling impairment in mouse cardiac myocytes. J Mol Cell Cardiol 2002. 34(10):1425-1431. [DOD] [CrossRef]
- Bucciarelli LG, Kaneko M, Ananthakrishnan R, Harja E, Lee LK, Hwang YC, Lerner S, Bakr S, Li Q, Lu Y, Song F, Qu W, Gomez T, Zou YS, Yan SF, Schmidt AM, Ramasamy R. Receptor for advanced-glycation end products: key modulator of myocardial ischemic injury. Circulation 2006. 113(9):1226-1234. [DOD] [CrossRef]
- Hudson BI, Bucciarelli LG, Wendt T, Sakaguchi T, Lalla E, Qu W, Lu Y, Lee L, Stern DM, Naka Y, Ramasamy R, Yan SD, Yan SF, D'Agati V, Schmidt AM. Blockade of receptor for advanced glycation endproducts: a new target for therapeutic intervention in diabetic complications and inflammatory disorders. Arch Biochem Biophys 2003. 419(1):80-88. [DOD] [CrossRef]
- Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000. 404(6779):787-790. [DOD] [CrossRef]
- Doria A, Nosadini R, Avogaro A, Fioretto P, Crepaldi G. Myocardial metabolism in type 1 diabetic patients without coronary artery disease. Diabet Med 1991. 8:S104-S107. [DOD]
- Bidasee KR, Zhang Y, Shao CH, Wang M, Patel KP, Dincer UD, Besch HR Jr. Diabetes increases formation of advanced glycation end products on Sarco(endo)plasmic reticulum Ca2+-ATPase. Diabetes 2004. 53(2):463-473. [DOD]
- Ren J, Bode AM. Altered cardiac excitation-contraction coupling in ventricular myocytes from spontaneously diabetic BB rats. Am J Physiol Heart Circ Physiol 2000. 279(1):H238-H244. [DOD]
- Iribarren C, Karter AJ, Go AS, Ferrara A, Liu JY, Sidney S, Selby JV. Glycemic control and heart failure among adult patients with diabetes. Circulation 2001. 103(22):2668-2673. [DOD]
- Kannel WB, McGee DL. Diabetes and cardiovascular disease. The Framingham study. JAMA 1979. 241(19):2035-2038. [DOD] [CrossRef]
- Fang ZY, Prins JB, Marwick TH. Diabetic cardiomyopathy: evidence, mechanisms, and therapeutic implications. Endocr Rev 2004. 25(4):543-567. [DOD] [CrossRef]
This article has been cited by other articles:

|
Emerging role of epigenetics and miRNA in diabetic cardiomyopathy
Asrih M, Steffens S
Cardiovasc Pathol 2012. Epub
|
|

|
Nox4-derived reactive oxygen species mediate cardiomyocyte injury in early type 1 diabetes
Maalouf RM, Eid AA, Gorin YC, Block K, Escobar GP, Bailey S, Abboud HE
Am J Physiol Cell Physiol 2012. 302(3):C597-C604
|
|
|
Particulate matter exposure exacerbates high glucose-induced cardiomyocyte dysfunction through ROS generation
Zuo L, Youtz DJ, Wold LE
PLoS One 2011. 6(8):e23116
|
|

|
Berberine attenuates cardiac dysfunction in hyperglycemic and hypercholesterolemic rats
Dong SF, Hong Y, Liu M, Hao YZ, Yu HS, Liu Y, Sun JN
Eur J Pharmacol 2011. 660(2-3):368-374
|
|

|
Potential role of nuclear factor kappaB in diabetic cardiomyopathy
Lorenzo O, Picatoste B, Ares-Carrasco S, Ramirez E, Egido J, Tunon J
Mediators Inflamm 2011. 2011:652097
|
|

|
Peroxisome proliferator-activated receptors, metabolic syndrome and cardiovascular disease
Azhar S
Future Cardiol 2010. 6(5):657-691
|
|

|
Maternal obesity induces fibrosis in fetal myocardium of sheep
Huang Y, Yan X, Zhao JX, Zhu MJ, McCormick RJ, Ford SP, Nathanielsz PW, Ren J, Du M
Am J Physiol Endocrinol Metab 2010. 299(6):E968-E975
|
|

|
The absence of pathological myofibre disarray in the diabetic heart: is it a paradox?
McLachlan CS, Lasker S, Keramat SM, Wang L, Jelinek H
Acta Cardiol 2009. 64(2):267-268
|
|

|
Shotgun Lipidomics of Neutral Lipids as an Enabling Technology for Elucidation of Lipid-Related Diseases
Gross RW, Han X
Am J Physiol Endocrinol Metab 2009. 297(2):E297-E303
|
|

|
Animal models of diabetes mellitus: relevance to vascular complications
Thompson CS
Curr Pharm Des 2008. 14(4):309-324
|
|

|
Cod liver oil supplementation improves cardiovascular and metabolic abnormalities in streptozotocin diabetic rats
Ceylan-Isik A, Hünkar T, Asan E, Kaymaz F, Ari N, Söylemezoglu T, Renda N, Soncul H, Bali M, Karasu C; The ADIC Study Group
J Pharm Pharmacol 2007. 59(12):1629-1641
|
|

|
Diabetic Cardiomyopathy in OVE26 Mice Shows Mitochondrial ROS Production and Divergence Between In Vivo and In Vitro Contractility
Song Y, Du Y, Prabhu SD, Epstein PN
Rev Diabet Stud 2007. 4(3):159-168
|
|
|


)
)
)

