Original Data
| Rev Diabet Stud,
2009,
6(1):37-42 |
DOI 10.1900/RDS.2009.6.37 |
Inflammation in Diabetic Encephalopathy is Prevented by C-Peptide
Anders A.F. Sima1,2, Weixian Zhang1, Christian W. Kreipke3, José A. Rafols3, William H. Hoffman4
1Department of Pathology, Wayne State University, Detroit, MI, USA
2Department of Neurology, Wayne State University, Detroit, MI, USA
3Department of Anatomy, Wayne State University, Detroit, MI, USA
4Department of Pediatrics, Medical College of Georgia, Augusta, GA, USA
Address correspondence to: Anders A.F. Sima, e-mail: asima@med.wayne.edu
Manuscript submitted May 18, 2009; resubmitted June 8, 2009; accepted June 9, 2009.
Keywords: type 1 diabetes, BB/Wor-rat, encephalopathy, inflammation, C-peptide, hippocampus, NF-kappaB, RAGE
Abstract
Encephalopathy is an increasingly recognized complication of type 1 diabetes. The underlying mechanisms are not well understood, although insulin deficiency has been implicated. The spontaneously diabetic BB/Wor-rat develops neuro-behavioral deficits and neuronal cell death in hippocampus and frontal cortex, which can be prevented by insulinomimetic C-peptide. Here we examined whether contributing factors such as activation of innate immune mediators are responsive to C-peptide replacement. Seven-month diabetic BB/Wor-rats and those treated with full C-peptide replacement were compared to age-matched control rats. Hippocampi of diabetic rats showed upregulation of RAGE and NF-κB, the former being localized to proliferating astrocytes. These changes were associated with increased expression of TNF-α, IL-1β, IL-2 and IL-6 in hippocampi of diabetic rats. Full C-peptide replacement, which did not induce hyperglycemia, resulted in significant prevention of upregulation of RAGE expression, activation of NF-κB and activation of pro-inflammatory factors. In conclusion, impaired insulin activity is associated with upregulation of RAGE and pro-inflammatory factors, and these are likely to contribute to previously described oxidative and apoptotic neuronal cell death. Replacement of insulinomimetic C-peptide significantly prevents this cascade of events.
Introduction
Diabetic encephalopathy is an increasingly recognized complication in type 1 diabetes [1]. It appears to be correlated with age of onset, duration of diabetes, and quality of glycemic control and leads to impaired learning abilities, memory and intelligence in affected patients [2, 3]. Experimental rodent models display impaired spatial learning and memory associated with impaired hippocampal long-term potentiation. These complications can be prevented by insulin treatment [4]. In long-term type 1 diabetic BB/Wor-rats, we previously demonstrated cognitive defects associated with apoptotic neuronal cell death in hippocampus and frontal cortex, changes that were prevented by pro-insulin C-peptide replacement [5]. Although the mechanisms underlying diabetic encephalopathy are not fully understood, the consequences of hyperglycemia and impaired insulin signaling are likely to play important pathophysiological roles [6-8]. Central insulin action has a modulating effect on the expression of other neurotrophic factors such as IGF-1 and NGF and their receptors; it inhibits oxidative stress-induced DNA damage by 8-OHdG and has significant beneficial effects on several caspases including active caspase 3 [5].
On the other hand, increased exposure of the brain to glucose and subsequent autoxidative glycosylation, oxidative-nitrosative stress and polyol-pathway products like carboxymethyl lysine (CML), pentosidine and fructose [9-13] are associated with increased accumulation of advanced glycation endproducts (AGE) and upregulation of its multi-ligand receptor RAGE, both in humans [7] and in experimental models of diabetes [8-10, 14, 15].
In this study, we examined the expression of RAGE and NF-κB and the associated activation of pro-inflammatory factors in hippocampi of chronically diabetic BB/Wor-rats and the preventive effect of pro-insulin C-peptide replacement.
Research design and methods
Animals
Fourteen pre-diabetic male BB/Wor-rats and seven age-matched non-diabetes prone BB/Wor-rats were obtained from Biomedical Research Models (Worcester, MA). Diabetes-prone rats developed diabetes at 73 ± 4 days of age and were given small daily substitution doses (0.5-3.2 IU) of protamine zinc insulin (Blue Ridge Pharma, Greensboro, NC) to maintain a steady hyperglycemic level of approximately 25 mmol/l and to prevent ketoacidosis (Table 1). Blood glucose levels were monitored weekly. The animals had free access to rat chow and drinking water. In seven diabetic animals, rat C-peptide (75 mmol × kg-1 × day-1, Sigma Genosys, The Woodlands, TX) was delivered by subcutaneously implanted osmopumps (Alzet, Palo Alto, CA) from the onset of diabetes throughout the observation period. At the time of sacrifice, after 7 months of diabetes, blood samples were collected from all animal groups for assessment of blood glucose, glycated hemoglobin levels, insulin and C-peptide levels.
Behavioral testing
The Morris water maze paradigm was used for behavioral testing and performed as originally described [16]. The testing of animals started two weeks prior to sacrifice. The animals were placed in a circular pool of water measuring 2.04 m in diameter and 0.4 m in height. The water temperature was kept at 28ºC. A platform was submerged 3 cm below the water, 10 cm from the pool's edge. The pool was divided into four quadrants (Q1-Q4). The animals were trained to reach the platform from the four quadrants for two consecutive days each week for two weeks prior to final testing. On each day they were given three acquisition trials. The final test was performed three days after the last training swim. The latencies to reach the platform from the four quadrants were recorded in seconds. Rats that failed to reach the platform in 100 s were lifted out and given a latency score of 100 s. Increased latencies reflect impaired learning and spatial memory.
Tissue collection
Animals were anesthetized and the whole brain was removed and bi-sected. One hemisphere was snap-frozen in liquid nitrogen and stored at -80ºC for protein isolation. The other hemisphere was fixed in buffered PBS (pH 7.4) 4% paraformaldehyde, paraffin embedded and 6 μm thick sections were prepared for immunohistochemistry.
Western blotting
Hippocampi were lysed in detergent lysin buffer (50 mmol/l Tris-HC1, pH 7.4, 150 mmol/l NaCl, 1 mmol/l EDTA, 1% Triton X-100, 1 mmol/l phenylmethylsulfonyl fluoride, 1 μg/ml leupeptin and 1 μg/ml aprotinin). Protein concentrations were measured using biocinchoninic acid protein assay reagent (Pierce, Rockford, IL). Ten to 40 μg was separated by 7.5-12% SDS-PAGE and transferred to polyvinylidine fluoride membranes (Bio-Rad, Hercules, CA). Membranes were blocked with Tween 20-Tris buffered saline containing 5% non-fat dry milk before incubation with primary antibodies. They were mouse anti-RAGE, mouse anti-NF-κB (Santa Cruz Biotechnology, Santa Cruz, CA), mouse anti-TNF-α and goat anti-IL-1β, anti-IL-2 (Sigma Aldrich, St. Louis, MO) and goat anti-IL6 (Pierce, Rockford, IL).
Antigen detection was performed using chemiluminescence (Amersham Pharmacia Biotech, Piscataway, NJ) with the appropriate horseradish peroxidase conjugated secondary antibodies. Membranes were exposed to Biomax film (Kodak, Rochester, NY) and densities were determined by Bio-Rad Fluoro-S multi-imager. Protein expression was corrected for actin expression.
Immunofluorescent immunostaining
Tissue sections were blocked with 10 mg/ml BSA and incubated with a mixture of rabbit anti-GFAP antibody (1:200, Sigma, MO) and mouse anti-RAGE antibodies (1:50, Santa Cruz) at room temperature for 1 hour at 4°C overnight. Sections were then washed in PBS and incubated with a mixture of FITC conjugated anti-rabbit secondary antibody (1:50, Sigma) and rhodamine conjugated anti-mouse secondary antibody (1:50, Chemicom, Teurecula, CA) at room temperature for 1 hour. The sections were mounted with Gel Mount (Biomedia, Foster City, CA) and examined using a Leica microscope (DMLB) equipped with SPOT RT image system and V 3.0 software (Diagnostic Instruments Inc, Sterling Heights, MI).
Statistical methods
All data are presented as means ± SD. The standard statistical package SPSS, V 13.0 was used to perform the analyses. Parametric statistics and analysis of variance (ANOVA) were used and Scheffes post hoc test was applied for group comparisons.
Results
Clinical data
Seven-month diabetic rats showed significant weight loss, hyperglycemia, elevated glycated hemoglobin, insulin deficiency and non-measurable C-peptide levels (all p < 0.001; Table 1). C-peptide treatment restored C-peptide levels to normal but did not alter body weight, glucose, glycated hemoglobin, or insulin levels in diabetic rats (Table 1).
Table
1.
Clinical data showing body weights, blood glucose levels, glycated Hb and plasma insulin and C-peptide concentrations in control, type 1 BB/Wor- and C-peptide replaced BB/Wor after 7 months of diabetes |
|
 |
 |
Legend:
* p < 0.001 vs. control rats. ** p < 0.001 vs. type 1 BB/Wor-rats. |
|
Behavioral studies
The latencies of diabetic BB/Wor-rats in the Morris water maze were significantly prolonged in the four quadrants (Figure 1). The latencies of C-peptide replaced diabetic rats were not significantly different from those of non-diabetic control rats in quadrants Q1, Q2 and Q4. Although the latency of C-peptide replaced diabetic rats in quadrant Q3 was significantly improved (p < 0.005), it remained prolonged (p < 0.01) compared to that of control rats (Figure 1).
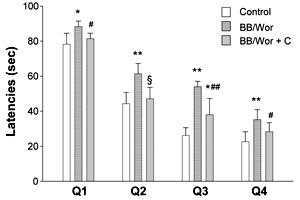 |
|
Figure 1. Results of behavioral tests. The figure shows latencies in seconds for controls, non-treated BB/Wor-rats and C-peptide replaced diabetic BB/Wor-rats in the four quadrants of the Morris water maze. * p < 0.01, ** p < 0.001 vs. control rats. # p < 0.05, ## p < 0.005, § p < 0.001 vs. untreated BB/Wor-rats. |
|
Immunocytochemistry
An increased number of astrocytes in hippocampal pyramidal cell layer, dentate fascia (Figure 2) and temporal white matter as identified by glial fibrillary acidic protein (GFAP) positivity was observed in diabetic animals. Immunopositive RAGE co-localized with GFAP in astrocytes (Figure 2) was increased in hippocampal pyramidal cells as well as in white matter oligodendrocytes.
C-peptide treatment prevented proliferation of GFAP-positive astrocytes and the increased immuno-expression of RAGE in hippocampal regions including the dentate fascia (Figure 2).
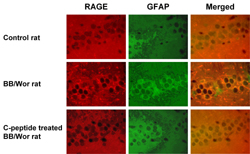 |
|
Figure 2. Immunocytochemistry of hippocampal dentate gyrus in control, diabetic and diabetic C-peptide-replaced animals. Increased staining for RAGE (red) and GFAP (green) indicates activation of astrocytes and increased RAGE expression. Merged images confirm the localization of RAGE mainly to astrocytes and their processes. Diabetic rats that received full replacement of rat C-peptide (Table 1) showed prevention of RAGE expression and only minimal GFAP positivity indicative of decreased astrocyte proliferation. Magnification 250X. |
|
Western blotting
The expression of NF-κB increased significantly in diabetic hippocampi (p < 0.01). This pattern was associated with increased expression of RAGE, TNF-α (both p < 0.01), IL-1β (p < 0.01) IL-2 and IL-6 (both p < 0.001) (Figure 3). Full C-peptide replacement in diabetic rats prevented significantly the upregulation of NF-κB, RAGE, TNF-α and IL-1β, IL-2 and IL-6 (Figure 3).
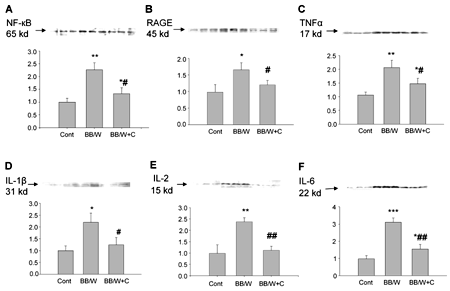 |
|
Figure 3. Immunoblot analyses of control, diabetic and C-peptide replaced diabetic hippocampi. NF-κB (A) was significantly upregulated in diabetic hippocampus, and this was significantly prevented by C-peptide. RAGE (B) was, as expected, significantly upregulated in diabetic rats and was mainly localized to proliferating astrocytes (cf. Figure 1). Significant prevention of RAGE upregulation by C-peptide in the presence of unaltered hyperglycemia was observed (cf. Table 1). TNF-α (C) expression was increased in diabetic rats and significantly but not fully prevented by C-peptide replacement. Additional pro-inflammatory factors IL-1β (D), IL-2 (E) and IL-6 (F) were upregulated in diabetic hippocampi, activations which were significantly prevented by C-peptide replenishment. * p < 0.05, ** p < 0.01, *** p < 0.001 vs. non-diabetic control rats. # p < 0.05, ## p < 0.01 vs. diabetic BB/Wor-rats. |
|
Discussion
In this study, we found that pro-inflammatory factors were upregulated in hippocampi of diabetic BB/Wor-rats and that replacement of insulinomimetic C-peptide prevented the upregulation of RAGE and NF-κB and the downstream activation of pro-inflammatory factors such as TNF-α, IL-1β, IL-2 and IL-6. These findings were associated with the prevention of proliferating astrocytes and significant prevention of deficits in learning and spatial memory as indicated by the Morris water maze paradigm.
AGE and nonenzymatically glycated or oxidated proteins, lipids or nucleic acids that are formed under conditions of oxidative stress and hyperglycemia [17] are traditionally regarded as the key activators of the multiligand RAGE receptor [18, 19]. However, in this study, C-peptide substitution had no effect on hyperglycemia, suggesting that other mechanisms are responsible for the upregulation of RAGE. We have previously demonstrated that C-peptide replacement has a beneficial effect on ROS-induced DNA damage in hippocampus in this model [5]. We suppose that this effect may provide an alternative source of RAGE upregulation. The most likely mechanism is that impaired insulin signaling, which is corrected by C-peptide [5, 20-22], directly or indirectly influences RAGE expression.
It has been shown that RAGE activation stimulates NF-κB [19, 23], although the exact signaling mechanism is not known [13]. An important consequence of NF-κB activation, which also occurs as a consequence of impaired insulin signaling [24-27], is the upregulation of RAGE itself [19, 28], hence providing a self-perpetuating loop. The mechanism underlying such an effect is that the gene encoding RAGE contains functional binding elements for NF-κB [29].
RAGE was localized to activated astrocytes, which along with microglia play key roles in the production of inflammatory mediators. RAGE is a multiligand receptor and has also been identified as a receptor for β-amyloid and prion fibrils [19]. Indeed, β-amyloid does accumulate in brains of BB/Wor-rats [30], and this potentially contributes to increased RAGE signaling in this model. The present data suggest that impaired insulin signaling per se may play a prominent role in RAGE signaling activities. This interpretation is supported by data obtained from diabetic mice treated with intranasal insulin, which does not effect systemic hyperglycemia [8]. Upregulation of RAGE has been described in the CNS in this mouse model [15]. In our study, we found prevention of CNS insulin signaling intermediaries, cognitive decline and structural changes. In the light of previous data [5, 14], these findings suggest a role of impaired insulin signaling in RAGE regulation and in diabetic encephalopathy.
Existing evidence supports the role of RAGE in innate immune responses [13, 15, 28]. Apart from promoting NF-κB activation, ligand interaction of RAGE activates MAPK family members like JNK, p38 and ERK1/2 [13, 31], thereby fueling the apoptogenic effects caused by aberration of the PI-3 kinase pathway [5]. It has been shown that interaction of RAGE with S100/calgranulin triggers macrophage activation through NF-κB resulting in the production of pro-inflammatory cytokines TNF-α and IL-1β [32].
The present findings showing an upregulation of several cytokines such as IL-2 and IL-6 is consistent with this cascade of signaling events [13, 33]. In addition to the already compromised hippocampal insulin receptor in the BB/Wor-rat [5, 34], activation of TNF-α inhibits Akt phosphorylation downstream of P13K in the insulin signaling pathway [35]. This inhibition is likely to impact on insulin's anti-apoptotic effects. It is therefore obvious that upregulation of RAGE signaling and activation of NF-κB result in a series of sometimes self-perpetuating events contributing to activation of inflammatory events and compromising anti-apoptotic regulators eventually resulting in apoptotic cell death in diabetic CNS [5, 14, 36].
Conclusions
In summary, the present data demonstrate that full substitution of insulinomimetic C-peptide in the type 1 diabetic BB/Wor-rat corrects the upregulation of RAGE and NF-κB activation with downstream beneficial effects on pro-inflammatory factors such as TNF-α and interleukins. These findings are associated with a preventative effect on the proliferation of RAGE-positive astrocytes in hippocampus and prevention of deficits in spatial memory and learning.
We conclude that activation of innate immune responses contributes to the development of diabetic encephalopathy and that it can be effectively prevented by replacement of insulinomimetic C-peptide.
Conflict of interest statement: The authors declare that they have no competing conflict of interests with respect to financial or other issues.
Acknowledgments:
This study was in part supported by grants from JDRF and the Thomas Foundation.
References
- Biessels GJ, Deary IJ, Ryan CM. Cognition and diabetes: a life span perspective. Lancet Neurol 2008. 7:184-190. [DOD] [CrossRef]
- Ferguson SC, Blane A, Wardlaw J, Frier BM, Perros P, McCrimman RJ, Deary IJ. Influence of early onset age of type 1 diabetes on cerebral structure and cognitive function. Diabetes Care 2005. 28:1431-1437. [DOD] [CrossRef]
- Schoenle EJ, Shoenle D, Molinari L, Largo RH. Impaired intellectual development in children with type 1 diabetes: association with HbA(1c), age of diagnosis and sex. Diabetologia 2002. 45:108-114. [DOD] [CrossRef]
- Biessels GJ, Kamal A, Urban IJ, Spruijt BM, Erkelens DW, Gispen WH. Water maze learning and hippocampal synaptic plasticity in streptozotocin-diabetic rats: effects of insulin treatment. Brain Res 1998. 800(1):125-135. [DOD] [CrossRef]
- Sima AA, Li ZG. The effect of C-peptide on cognitive dysfunction and hippocampal apoptosis in type 1 diabetes. Diabetes. 2005. 54:1497-1505. [DOD]
- Perantie DC, Wu J, Koller JM, Lim A, Warren SL, Black KJ, Sadler M, White NH, Hershey T. Regional brain volume differences associated with hyperglycemia and severe hypoglycemia in youth with type 1 diabetes. Diabetes Care 2007. 30:2331-2337. [DOD] [CrossRef]
- Hoffman WH, Artlett CM, Zhang W, Kreipke CW, Passmore GG, Rafols JA, Sima AA. Receptor for advanced glycation end products and neuronal deficit in fatal brain edema of diabetic ketoacidosis. Brain Res 2008. 1238:154-162. [DOD] [CrossRef]
- Francis GJ, Martinez JA, Liu WQ, Xu K, Ayer A, Fine J, Tuor UI, Glazner G, Hanson LR, Frey WH 2nd, Toth C. Intranasal insulin prevents cognitive decline, cerebral atrophy and white matter changes in murine type I diabetic encephalopathy. Brain 2008. 131(Pt 12):3311-3334. [DOD] [CrossRef]
- Requena JR, Baynes JW. Studies in animal models on the role of glycation and advanced glycation end-products (AGE's) in the pathogenesis of diabetic complications: pitfalls and limitations. In: Sima AAF (ed). Chronic Complications in Diabetes. Harwood Acad Publ, Amsterdam 2000, pp 43-70. [DOD]
- Schalkwijk CG, Stehouwer CD, Hinsbergh VW. Fructose-mediated non-enzymatic glycation: sweet coupling or bad modification. Diab Metab Res Rev 2004. 20:369-382. [DOD] [CrossRef]
- Kuhad A, Bishnoi M, TiWari V, Chopra K. Suppression of NF-kappa beta signaling pathway by tocotrienol can prevent diabetes associated cognitive deficits. Pharmacol Biochem Behav 2009. 92:251-259. [DOD] [CrossRef]
- Kamboj SS, Chopra K, Sandhir R. Neuroprotective effect of N-acetylcystein in the development of diabetic encephalopathy in streptozotocin-induced diabetes. Brain Dis 2008. 23:427-443. [DOD] [CrossRef]
- Lin L. RAGE on the toll road? Cell Mol Immunol 2006. 3:351-358. [DOD]
- Sima AA, Zhang W, Hoffman WH. Apoptosis of oligodendroglia cells contributes to white matter changes in type 1 diabetic encephalopathy. Proc Neurodiab XVIII 2008. 94. [DOD]
- Toth C, Schmidt AM, Tuor UI, Francis G, Foniok T, Brusse V, Kaur J, Yan SF, Martinez JA, Barber PA, Cuchan A, Zochodne DW. Diabetes, leucoencephalopathy and rage. Neurobiol Dis 2006. 23:445-461. [DOD] [CrossRef]
- Morris R. Development of a water maze procedure for studying spatial learning in the rat. J Neurosci 1984. 11:47-60. [DOD]
- Ahmed N, Ahmed U, Thornalley PJ, Hager K, Fleischer G, Munch G. Protein glycation, oxidation and nitration adduct residues and free adducts of cerebrospinal fluid in Alzheimer's disease and link to cognitive impairment. J Neurochem 2005. 92:255-263. [DOD] [CrossRef]
- Neeper M, Schmidt AM, Brett J, Jan SD, Wang F, Pan YL, Elliston K, Stern D, Shaw A. Cloning and expression of RAGE: a cell surface receptor for advanced glycosylation end products of proteins. J Biol Chem 1992. 267:14998-15004. [DOD]
- Ramasamy R, Vanucci SJ, Yan SD, Herold K, Yan SF, Schmidt AM. Advanced glycation end products and RAGE: a common thread in aging, diabetes, neurodegeneration and inflammation. Glycobiology 2004. 15:16R-28R. [DOD] [CrossRef]
- Grunberger G, Qiang X, Li ZG, Mathews ST, Sbriessa D, Shisheva A, Sima AA. Molecular basis for the insulinomimetic effects of C-peptide. Diabetologia 2001. 44:1247-1257. [DOD] [CrossRef]
- Li ZG, Qiang X, Sima AA, Grunberger G. C-peptide attenuates protein tyrosine phosphatase activity and enhances glycogen synthesis in L6 myoblasts. Biochem Biophys Res Com 2001. 26:615-619. [DOD]
- Sima AA, Zhang W, Li ZG, Murakawa Y, Pierson CR. Molecular alterations underlie nodal and paranodal degeneration in type 1 diabetic neuropathy and are prevented by C-peptide. Diabetes 2004. 53:1556-1563. [DOD] [CrossRef]
- Yan SD, Chen X, Schmidt AM, Brett J, Godman G, Scott CW, Caputo C, Frappier T, Yen SH, Stein D. Glycated tau in Alzheimer disease: a mechanism for induction of oxidant stress. Proc Natl Acad Sci USA 1994. 91(16):7787-7791. [DOD] [CrossRef]
- Li ZG, Zhang W, Sima AA. C-peptide enhances insulin-mediated cell growth and protection against high glucose induced apoptosis in SH-SY5Y cells. Diab Metab Res Rev 2003. 19:375-385. [DOD] [CrossRef]
- Hayden MS, Shosh S. Signaling to NF-kappaB. Genes Dev 2004. 18(18):2195-2224. [DOD] [CrossRef]
- Cifarelli V, Luppi P, Tse HM, He J, Piganelli J, Trucco M. Human proinsulin C-peptide reduces high glucose-induced proliferation and NF-kappaB activation in vascular smooth muscle cells. Atherosclerosis 2008. 201:248-257. [DOD] [CrossRef]
- Bierhaus A, Schiekofer S, Schwarninger M, Andrassy M, Humpert PM, Chen J, Hong M, Luther T, Henle T, Klöting I, Morcos M, Hoffman M, Tritschler H, Weigle B, Kasper M, Smith M, Perry G, Schmidt AM, Stern DM, Häring HU, Schleicher E, Nawroth PP. Diabetes-associated sustained activation of the transcription factor nuclear factor-kappaB. Diabetes 2001. 50:2792-2808. [DOD] [CrossRef]
- Mukherjee TK, Mukhopadhyay S, Hoidal JR. The role of reactive oxygen species in TNF alpha-dependent expression of the receptor for advanced glycation end products in human umbilical vein endothelial cells. Biochim Biophys Acta 2005. 1744:213-223. [DOD] [CrossRef]
- Li J, Schmidt AM. Characterization and functional analysis of the promotor of RAGE, the receptor for advanced glycation end products. J Biol Chem 1997. 272:16498-16506. [DOD] [CrossRef]
- Li ZG, Zhang W, Sima AA. Alzheimer-like changes in rat models of spontaneous diabetes. Diabetes 2007. 56:1817-1824. [DOD] [CrossRef]
- Stein D, Yan SD, Yan SF, Schmidt AM. Receptor for advanced glycation end products: a multiligand receptor magnifying cell stress in diverse pathologic settings. Adv Drug Deliv Rev 2002. 54:1615-1625. [DOD] [CrossRef]
- Hoffman MA, Drury S, Fu C, Qu W, Tagushi A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P, Neurath MF, Slattery T, Beach D, McClary J, Nagashima M, Morser J, Stern D, Schmidt AM. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell 1999. 97:889-901. [DOD] [CrossRef]
- Karin M. Inflammation-activated protein kinases as targets for drug development. Proc Am Thorac Soc 2005. 2:380-390. [DOD]
- Sima AA, Kamiya H, Li ZG. Insulin, C-peptide, hyperglycemia and central nervous system complications in diabetes. Eur J Pharmacol 2004. 490(1-3):187-197. [DOD] [CrossRef]
- Goetze S, Blaschke F, Stawowy P, Bruemmer D, Spencer C, Graf K, Gräfe M, Law RE, Fleck E. TNF-alpha inhibits insulin's anti-apoptotic signaling in vascular smooth muscle cells. Biochem Biophys Res Commun 2001. 287:662-670. [DOD] [CrossRef]
- Li ZG, Zhang W, Sima AA. The role of impaired insulin/IGF action in primary diabetic encephalopathy. Brain Res 2005. 1037:12-24. [DOD] [CrossRef]
This article has been cited by other articles:

|
C-peptide reduces high-glucose-induced apoptosis of endothelial cells and decreases NAD(P)H-oxidase reactive oxygen species generation in human aortic endothelial cells
Cifarelli V, Geng X, Styche A, Lakomy R, Trucco M, Luppi P
Diabetologia 2011. 54(10):2702-2712
|
|

|
Early transcriptional regulation by C-peptide in freshly isolated rat proximal tubular cells
Lindahl E, Nordquist L, Müller P, El Agha E, Friederich M, Dahlman-Wright K, Palm F, Jörnvall H
Diabetes Metab Res Rev 2011. 27(7):697-704
|
|

|
Oxidative damage is present in the fatal brain edema of diabetic ketoacidosis
Hoffman WH, Siedlak SL, Wang Y, Castellani RJ, Smith MA
Brain Res 2011. 1369:194-202
|
|

|
C-peptide and long-term complications of diabetes
Luppi P, Cifarelli V, Wahren J
Pediatr Diabetes 2011. 12(3Pt2):276-292
|
|

|
Anti-inflammatory effects of C-peptide prevent endothelial dysfunction in type 1 diabetes
Cifarelli V, Trucco M, Luppi P
Immunol Endocr Metab Agents Med Chem 2011. 11(1):59-70
|
|

|
Encephalopathies: the emerging diabetic complications
Sima AA
Acta Diabetol 2010. 47(4):279-293
|
|

|
Insulin and IGF-1 receptors, nitrotyrosin and cerebral neuronal deficits in two young patients with diabetic ketoacidosis and fatal brain edema
Hoffman WH, Andjelkovic AV, Zhang W, Passmore GG, Sima AA
Brain Res 2010. 1343:168-177
|
|

|
Neuroprotection in diabetic encephalopathy
Fazeli SA
Neurodegener Dis 2009. 6(5-6):213-218
|
|

|
Sequential abnormalities in type 1 diabetic encephalopathy and the effects of C-Peptide
Sima AA, Zhang W, Muzik O, Kreipke CW, Rafols JA, Hoffman WH
Rev Diabet Stud 2009. 6(3):211-222
|
|

|
The beneficial effects of C-Peptide on diabetic polyneuropathy
Kamiya H, Zhang W, Sima AA
Rev Diabet Stud 2009. 6(3):187-202
|
|

|
Anti-inflammatory properties of C-Peptide
Haidet J, Cifarelli V, Trucco M, Luppi P
Rev Diabet Stud 2009. 6(3):168-179
|
|

|
Breakthrough in diabetes therapy ... Just around the corner?
Rudert WA, Trucco M
Rev Diabet Stud 2009. 6(2):76-80
|
|
|


)
)
)
)

