Chapter V. Beta-Cell Replacement
| Rev Diabet Stud,
2012,
9(4):407-416 |
DOI 10.1900/RDS.2012.9.407 |
Islet Neogenesis: A Possible Pathway for Beta-Cell Replenishment
Susan Bonner-Weir, Lili Guo, Wan-Chun Li, Limor Ouziel-Yahalom, Philippe A. Lysy, Gordon C. Weir, Arun Sharma
Joslin Diabetes Center, Department of Medicine, Harvard Medical School, 1 Joslin Place, Boston, MA 02215
Address correspondence to: Susan Bonner-Weir, e-mail susan.bonner-weir@joslin.harvard.edu
Manuscript submitted December 16, 2012; accepted January 21, 2013.
Keywords: diabetes, islet, insulin, pancreatic beta-cell, neogenesis, regeneration, replication
Abstract
Diabetes, particularly type 1 diabetes, results from the lack of pancreatic β-cells. β-cell replenishment can functionally reverse diabetes, but two critical challenges face the field: 1. protection of the new β-cells from autoimmunity and allorejection, and 2. development of β-cells that are readily available and reliably functional. This chapter will examine the potential of endogenous replenishment of pancreatic β-cells as a possible therapeutic tool if autoimmunity could be blunted. Two pathways for endogenous replenishment exist in the pancreas: replication and neogenesis, defined as the formation of new islet cells from pancreatic progenitor/stem cells. These pathways of β-cell expansion are not mutually exclusive and both occur in embryonic development, in postnatal growth, and in response to some injuries. Since the β-cell population is dramatically reduced in the pancreas of type 1 diabetes patients, with only a small fraction of the β-cells surviving years after onset, replication of preexisting β-cells would not be a reasonable start for replenishment. However, induction of neogenesis could provide a starting population that could be further expanded by replication. It is widely accepted that neogenesis occurs in the initial embryonic formation of the endocrine pancreas, but its occurrence anytime after birth has become controversial because of discordant data from lineage tracing experiments. However, the concept was built upon many observations from different models and species over many years. Herein, we discuss the role of neogenesis in normal growth and regeneration, as learned from rodent models, followed by an analysis of what has been found in humans.
Abbreviations: BrdU – bromodeoxyuridine; CAII – carbonic anhydrase II; CCKB – cholecystokininB; CK – cytokeratin; CT – computed tomography; ERT2 – estrogen receptor T2; FoxM1 – forkhead box protein M1; GFP – green fluorescent protein; GLP-1 – glucagons-like peptide 1; HNF1β – hepatocyte nuclear factor 1 beta; Ins – insulin; MAFA – v-maf musculoaponeurotic fibrosarcoma oncogene homolog A (avian); MIP – mouse insulin I gene promoter; PAX4 – paired box gene 4; PDL - partial duct ligation; PDX1 – pancreatic and duodenal homeobox 1; Px – partial pancreatectomy; SOX9 – SRY-box 9; SRY – sex-determining region Y; T2D – type 2 diabetes; TGFα – transforming growth factor alpha
1. Introduction
Type 1 diabetes results from a marked deficiency of pancreatic β-cells, while type 2 diabetes has only a relative deficiency. The Edmonton protocol for islet transplantation has provided the "proof-of-concept" that β-cell replenishment can reverse type1 diabetes. However, the field still has two critical challenges: 1. the protection of β-cells from autoimmunity and allorejection, and 2. the provision of β-cells that are readily available and reliably functional. The area of β-cell replenishment or replacement is a major area of research. While another chapter in this Special Issue deals with islet transplantation as an exogenous supply of β-cell replacement [1], this chapter will examine the endogenous replenishment of pancreatic β-cells.
Normal growth of the endocrine pancreas relies on differentiation of islet cells from stem cells/progenitors (a process termed neogenesis) and on replication of preexisting islet cells. Replication and neogenesis are not mutually exclusive; both pathways of β-cell expansion occur in embryonic development, in postnatal growth, and in response to some injuries. In 1938, the pathologist Shields Warren wrote: "The pancreas in diabetes is not simply the scarred field of an old battleground, but is the actual field of conflict. It does not submit without a struggle to injury, but endeavors to regenerate" [2]. Yet, by several years after onset, the pancreas in type 1 diabetes usually has very few surviving β-cells, so just enhancing sufficient replication of these remaining β-cells to reach a critical mass is a difficult and perhaps unreachable goal, even if the autoimmune destruction could be controlled. However, enhancing neogenesis may have more potential to provide an increase of new β-cells that could then replicate further to provide enough β-cells to reverse diabetes.
2. Definition of neogenesis
Neogenesis or the formation of new islet cells from pancreatic progenitor/stem cells is widely accepted as being responsible for the initial embryonic formation of the endocrine pancreas. However, its occurrence after birth has become controversial because of discordant observations in lineage tracing experiments. The concept was built upon numerous observations from different models and species over many years, and there are compelling data that progenitors have a role in renewal and growth of islets after birth.
One of the difficulties in measuring the extend of neogenesis has been the lack of clear identifying markers of such newly formed cells. The most common criteria for identification have been islet hormone-positive cells that seem to be budding from the pancreatic duct epithelium, or tiny clusters (1-3 cells) of scattered insulin-positive cells (Figure 1). It has been argued that these cells are static without actual "budding", and that they are relics from embryonic development. Nonetheless, many rodent studies, including some lineage tracing experiments, provide data on neogenesis that accompanies normal growth. Additional data come from surgical and experimental manipulations. These data support the concept of neogenesis being a dynamic process. Other rodent studies have shown an increase in the proportion of small islets or even a reduced mean islet size at the time of islet expansion that supports the formation of new islets after birth and injury. Some of these animal data will be presented in this perspective overview, followed by an analysis of the data from human studies.
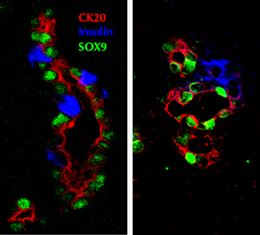 |
 |
Figure 1. Immunostaining of neogenesis. The figure shows two images of regenerating areas in the pancreas 4 days after partial pancreatectomy in the young adult rat. Insulin-positive (blue) cells are budding from the pancreatic ducts (cytokeratin 20: red, Sox9: green). These cells are newly differentiated from progenitors within the ducts as these focal areas could only be seen at 48 hr after surgery. |
|
3. Neogenesis during the neonatal period in rodents
During the neonatal period, β-cell replication continues, and significant neogenesis occurs. There have been numerous approaches that have supported the significance of the neogeneic formation during the neonatal period in both rats and mice. In a mathematical model based on existing data on determinants of β-cell mass (mass, individual cell volume, replication, and apoptosis) in Sprague-Dawley rats [3], we predicted two waves of neogenesis: one immediately after birth and the other 2-3 weeks after birth. We subsequently documented the increased appearance of islets budding from the ducts at the same times, shortly after birth and just before weaning [4], supporting the predicted waves of neogenesis. Using data from the latter study, we estimated that about 70% of the β-cells seen at day 31 could be accounted for by replication of pre-existing β-cells, while the remaining 30% came from neogenesis [5]. This calculated estimate is consistent with the findings from our duct-specific lineage tracing experiments in mice ([6]; Guo et al., unpublished). Similarly, Bouwens et al. showed that the β-cell mass in rats more than doubled between 2 and 5 days after birth but the bromodeoxyuridine (BrdU) incorporation in β-cells at day 2 could account for only 12% of the observed growth by day 5. No change in the cross-sectional area of the β-cells accompanied the increase in mass, indicating that new cells rather than larger cells accounted for the increased mass. The authors concluded that most of the growth was by neogenesis occurring at the periphery of the islets from cells strongly expressing cytokeratin (CK) 19 and 20 [7].
Using measurements of islet size, number and β-cell proliferation, several groups have provided documentation of a substantially increased number of islets or insulin-positive cell clusters occurring from birth to adulthood. After extensive counting of total β-cells at day 2-3 and 10 weeks, Chintinne et al. concluded that about 30,000 β-cell clusters of less than 50 mm diameter formed during this time in male Wistar rats [8]; this represented a 14-fold increase in the number of small aggregates of insulin-positive cells. Similarly, Peng et al. showed that the islet number in mice increased from about 50/pancreas at 1 week to about 900 at 2 months after which it was stable, suggesting that impressive neogenesis occurred after birth, but became undetectable by 2 months [9]. An obvious caveat for the enumeration of islets over time is that small clusters of islet cells, which initially had been below the detection limit, may increase in size due to proliferation and be counted as newly formed. However, the former counted individual cells, and the latter used a cutoff of 20 μm, which is essentially the diameter of 2 cells. An alternative interpretation to new formation of islets during this neonatal time is that of fission of large islets [10]. Using mice that express green fluorescent protein (GFP) under the control of the mouse insulin I gene promoter (MIP), MIP-GFP CD1 mice, and a criteria of greater than 4 insulin-positive cells per islet, Jo et al. presented a model based on islet diameter of fission of larger elongated islets during the first 3 weeks after birth [11]. Over the first 3 postnatal weeks they estimated, 800 new islets were formed, 300 of which formed by fission and 500 by neogenesis.
A third approach has been lineage tracing using the Cre-lox system. Several different promoters used as duct-specific drivers of Cre expression in crossbreeds with reporter mice gave differing results, with the following having similar conclusions:
1. Using Hnf1β promoter, Solar et al. found no marked β-cells or acinar cells after birth [12]
2. Using mucin1, Kopinke et al. also found no neonatal neogenesis, but only had a 10% labeling efficiency in ducts [13]
3. Kopp et al. [14] and Furuyama et al. [15], both using Sox9 promoter, found a low rate of neogenesis only immediately after birth, with the former finding absolutely no acinar-labeling, while the latter found acinar-labeling such that by 1 year most acinar cells were marked.
In contrast, using carbonic anhydrase II (CAII), which only starts to be expressed in pancreatic ducts at the very end of gestation [16], we showed that in the 4 weeks after birth both islets and acini were formed from cells that once expressed CAII; 38% of the islets were marked (17% of all insulin-positive cells), and a number of acinar cells with some lobes were marked, but not others [6]. In an effort to reconcile these discrepant results, we hypothesized that the duct cells are heterogeneous in their transcription factor expression and their potential to act as progenitors for islets and acini. We titrated the HNF1β and SOX9 antibodies in immunofluorescent staining to determine whether there were heterogeneous protein levels of these transcription factors. Indeed, different expression levels of HNF1β and SOX9 were found in cells through the ductal tree in both human and mouse adult pancreas [17]. The significance of the heterogeneity of the pancreatic duct transcriptional profile and the resultant differences in progenitor potential may explain the discrepant findings of lineage tracing. These processes are currently being studied.
Lineage tracing using the insulin2 promoter as driver and looking for dilution of the label during chase periods initially sparked the controversy over neogenesis [18]. No evidence of dilution of the genetic marking of β-cells in rat insulin promoter:CreER (Cre recombinase estrogen receptor T2) mice was found either in adults or after partial pancreatectomy, leading Dor et al. to conclude that replication was the mechanism of β-cell expansion in adult mice and neogenesis did not occur. Their approach of measuring dilution of marked cells is complicated by a possible leakiness of the Cre-lox system over time and the retention of tamoxifen for weeks that could have led to misleading conclusions [19, 20]. The tamoxifen doses commonly used to induce Cre-loxP recombination were shown in transplantation experiments to continue to label significant numbers of cells for weeks after tamoxifen treatment [20]. Additionally, the negative result in Dor et al. may be caused by their failure to examine the neonatal period or the new lobes after pancreatectomy (see below). Using the same approach, Nakamura et al. recently examined the neonatal period and reported that neogenesis occurred in the second to third week after birth, but not in adults [21].
4. Experimental models of neogenesis
The classic rodent models of pancreatic regeneration, partial pancreatectomy [22] and partial duct ligation [23], have provided evidence for both increased β-cell replication and neogenesis. Other experimental treatments, including dietary soybean trypsin inhibitor [24], GLP-1 receptor agonists [25], betacellulin [26], and cellophane wrapping of the head of the pancreas (a partial duct obstruction) [27] have been reported to induce neogenesis. Additional strong evidence of formation of new β-cells by neogenesis comes from a number of transgenic models, including overexpression of interferon-γ in the β-cells [28], overexpression of transforming growth factor (TGF) α in pancreatic ducts [29], and Pax4 ectopic expression in glucagon-positive cells [30]. In the latter, β-cells are thought to originate from an α-cell lineage rather than a duct cell lineage.
4.1 Partially pancreatectomized rodents
Partial pancreatectomy (Px, 90% in rats, 60-80% in mice) has resulted in substantial regeneration of the adult pancreas with whole new lobes containing islets being formed accompanied by enhanced proliferation of pre-existing β and acinar cells [22, 31-33]. Regeneration is not limited to young animals; both replication and neogenesis could be demonstrated in retired breeder rats (500 g) after 90% Px [34]. The partial Px model has been applied to mice using less extensive resection, often only 50%. At least two groups, using 60% Px in adult mice, reported neogenesis and enhanced replication [35, 36], while others reported enhanced replication only [18, 37, 38]. Their lack of observed neogenesis may be due to a lesser extent of resection of the pancreas or to the fact that the newly regenerated pancreatic lobes were not studied. In our own studies, we found less regeneration with less resection [39].
We have studied the 90% Px adult rat extensively. In this model, the well-defined 10% remnant regenerated, reaching 27% of the sham pancreatic weight and 45% β-cell mass of sham-operated rats by 8 wks [22, 25]. While β-cell replication and hypertrophy contributed to this increased mass, so did neogenesis. There was an expansion of the ductal tree, starting at the common pancreatic duct, with subsequent appearance of branching ductules in well-defined areas (foci of regeneration) that developed into new pancreatic lobes [22, 40, 41]. In this process, the mature pancreatic duct cells rapidly lost expression of key markers of the mature duct phenotype and replicated [40]. At least, some duct cells regressed to a progenitor-like phenotype, and then appeared to redifferentiate through a pathway used during normal embryonic development, transiently expressing PDX1 protein [41] and the cascade of transcription factors seen in embryonic development, including transient expression of the endocrine lineage-specific transcription factor neurogenin 3 [40]. As the focal areas of regeneration differentiated into new pancreatic lobes, the islets within these areas resembled embryonic islets with a higher proportion of insulin-positive cells lacking MAFA expression. The proportion of MAFA+ insulin-positive cells increased as foci further differentiated, but even in mature foci the proportion of MAFA-expressing β-cells was lower than in the remnant pancreas of the same animals [40]. Interestingly, new lobes of the pancreas may even form spontaneously in normal adult animals; occasional pancreatic lobes with high BrdU incorporation in all cell types were seen in 6-month old rats when most of the pancreas had almost none [3, 5].
4.2 Partial duct ligation in rodents
The partial duct ligation model used for several decades in rats [23, 42-45] has more recently been transferred to mice. In a comprehensive study in rats, Wang et al. reported that, distal to the ligation, the β-cell population doubled in one week with increased numbers of small islets and islet cell clusters [23]. Since the β-cell labeling index (BrdU incorporation) could not account for the increase, they concluded that islet neogenesis had occurred. Using this rat model, gastrin was shown to stimulate β-cell neogenesis in the distal portion, but not in the pancreas proximal to the ligation. This effect occurred as the cholecystokinin B (CCKB) receptor, through which gastrin functions, was induced only in the distal portion [45].
With the ability to do lineage tracing in mice using the Cre-lox system, the partial duct ligation (PDL) model has been recently used extensively, but with conflicting results. Neogenesis after PDL was reported in several studies [6, 46], but in others no marked β-cells were found after PDL [12, 14]. Using neurogenin 3 reporter mice, Heimberg's group showed that there was an induction of neurogenin 3+ cells after partial duct ligation [46]. These cells arose in or adjacent to the pancreatic ducts and differentiated into islet cells in explants of embryonic pancreas from neurogenin 3null mice. Using SOX9 CreERT2 driver, Kopp et al. found enhanced neurogenin 3 expression after PDL, but with no labeled β-cells, suggesting that the Sox9-labeled duct cells could start endocrine differentiation, but may not have completed it [14]. Chintinne et al. suggested that this discrepancy may be due to the limited amount of islet regeneration in this model [47]. They concluded that PDL resulted in neogenesis, with a 2.2-fold increase in small β-cell clusters that were highly proliferative, but that the number of the proliferative β-cells accounted for less than 5% of the total β-cell mass at 2 weeks post PDL.
Using Elastase (acinar-specific) or Pdx1 (global pancreas)-driven expression of Cre to target diphtheria toxin ablation of the pancreatic tissue, Criscimanna et al. showed that the severity of injury may determine the regenerative mechanism, and that in adult mice both new acinar and islet cells could arise from ductal cells [48].
4.3 Selected examples of other transgenic mice with induced neogenesis
Besides the use of reporter mice and the Cre-lox system for lineage tracing, transgenic mice with overexpression of various cytokines or growth factors have provided evidence of neogenesis. Studies were performed by the Sarvetnick group on the human insulin promoter using interferon-γ transgenic mice. The group found continual inflammatory destruction of islets associated with continued proliferation of ductal epithelium and budding of new islets from the ducts [49]. The ductal expression of PDX1 protein and other transcription factors seen in embryonic pancreatic development suggested that this regeneration recapitulated the embryonic process [50, 51]. Similarly, enhanced PDX-1 expression (as might be expected with replicated duct cells) and focal expression of Pax6 [52] were found within the metaplastic ducts after induction of TGF-α in the exocrine pancreas in the metallothionein1-TGF-α mouse [53]. Also, 6% of the duct cells were immunostained for insulin [29], supporting the hypothesis of ongoing islet neogenesis. When metallothionein1-TGF-α mice were crossed with mice transgenic for rat insulin promoter driving gastrin expression, the metaplastic ducts were extensively reduced and islet mass increased, suggesting gastrin as a factor that could drive differentiation of progenitors to islets [29].
In mice in which β-cell replication was blocked by pancreatic deletion of FoxM1, compensatory growth of the β-cell mass did not occur during pregnancy, but normal β-cell mass was restored in the early postpartum period. This restoration was suggested to occur by neogenesis, as indicated by the increased number of endocrine clusters, increased proportion of small islets, and increased neurogenin 3+ cells adjacent to the ducts [54]. A similar case of induction of neogenesis was seen in transgenic mice in which Pax4 was expressed in glucagon-expressing α-cells [30]. The ectopic expression of Pax4 reprogrammed α-cells to insulin-producing β-cells, yet there was continual renewal of α-cells. The authors concluded that there was continued α-cell neogenesis in these mice. This conclusion was based on the evidence of replicating duct cells, rather than of hormone-positive cells, of labeling of hormone-positive cells after lentiviral reporter administered through the ductal tree only in the transgenic mice and not control mice, and of blockage of the replenishment of α-cells by a knockdown of neurogenin 3 [30].
If ductal cells gave rise to new endocrine cells after birth, one might expect some residual expression of duct markers in the newly formed cells. Such transient expression of duct cell markers has been reported in β-cells of newborn rats [55], in regenerated islets after partial pancreatectomy [5, 56], in grafts of purified human duct cells [57], and in islets of mice conditionally expressing Pax4 in glucagon-producing cells [30]. These observations suggest the recent passage of the new cells through the ductal phenotype.
5. Postnatal expansion of the endocrine pancreas in humans
Unlike the experimental approaches that one can use in rodents, our knowledge of the growth of the human endocrine pancreas is limited to a few studies on autopsied pancreas. In a study of 46 autopsied pancreatic organs from children, the Butler group [58] reported the fractional insulin-positive area was 2.6 ± 0.5% pancreatic area between birth and 2 yr and 1.3 ± 0.2% at 18.5-21.5 yr. This decrease in the fractional area (relative volume or volume density) coincides with the approximate 5-fold expansion of the pancreas volume shown in a comprehensive study of the pancreas organs of living humans using computed tomography (CT) [59]. The pancreatic parenchyma was estimated to be less than 10 cm3 in children under the age of 5, with growth to about 50 cm3 by age 20, then stable levels for a couple of decades, followed by a gradual decrease with age. However, there is a wide variation in parenchymal volume at any age, with 17-50 cm3 at age 11-13 years (n = 11) and 30-73 cm3 at age 19-21 years (n = 8). Even though this wide variation limits the accuracy of using these data for estimating β-cell mass in other children, the lack of other more accurate data led the Butler group to use an equation derived from these data for estimating pancreatic volume and then β-cell mass (pancreatic volume X relative density of β-cells) [58].
Their conclusions were initially surprising. While the β-cell mass expanded several times from birth to adulthood, it occurred by increasing islet size through replication of preexisting β-cells rather than through an increasing number of islets, with most of the growth occurring before the age of 5 and no secondary spurt of growth during adolescence. Similarly, a recent study by Gregg et al. on 30 autopsied pancreas organs from children from birth to 18 years also found little β-cell growth after 2 yr age [60]. However, in contrast to the Butler group study, they found no increased mean islet size from birth to older adult. Additionally, their measures of neogenesis (CK19+ insulin-positive cells and insulin-positive cells within the duct epithelium) had small increases between 2 and 7 years and again in adults.
5.1 Evidence of compensatory expansion in the adult human pancreas
In studies on autopsied adult pancreas organs, there seems to be some compensatory growth of the β-cells in case of obesity [61-66]. For example, the fractional area of β-cells (% insulin area/pancreatic area) was 1.7 ± 0.3% for lean non-diabetics and 2.6 ± 0.4% for obese non-diabetics in a Minnesota population [62]. From rodent models, we learned that compensatory elevations in β-cell mass can result from increased proliferation (hyperplasia), individual cell volume increase (hypertrophy), and new differentiation from stem/progenitor cells. Yet, β-cell proliferation in adult humans has been reported as extremely low, and greatly enlarged islets are rarely found. In autopsied human pancreata, β-cell replication (Ki67+ β-cells) drops to less than 0.2% already by 5 years of age [58], in another study to about 0.5% in adolescents [60], and can be almost negligible in adults [60, 62, 67, 68]. However, this low level of detection may result from the tissue being only retrieved after death. In a recent study on human organ donor pancreata, all pancreata had Ki67+ β-cells, with a stable, albeit low, percentage in donors from 40 to 65 years of age [69]. In another study, donor pancreata had little or no Ki67+ insulin-positive cells, but grafts of isolated islets from such donors had 0.22 ± 0.03% Ki67+ insulin-positive cells after 4 week transplantation in immunocompromised normoglycemic mice; this level was also found in frozen human pancreas obtained at surgery [70]. An important concept recently emphasized by Chintinne et al. in rodents is that, in the adult, the large β-cell population can provide a significant pool of proliferating β-cells even though the proliferation frequency (% Ki67+ cells) is lower than in neonates [8].
Using insulin-positive cells found in the ductal epithelium, insulin-positive CK+ cells, and small clusters of 1-3 insulin-positive cells scattered in the parenchyma as identifiers of neogenetic islets, active neogenesis in the adult human pancreas is suggested by a number of studies (Table 1). In adults, the frequencies reported are in the range of 0.4-0.6% insulin-positive duct cells whether in organ donors or autopsied pancreas, although in some pancreata no insulin-positive cells were detectable [58, 69, 71, 72]. The dynamic nature of these insulin-positive cells is suggested by their increase seen in pancreas organs from obese patients [71, 72], in patients with chronic pancreatitis [73] during pregnancy [74], and in patients receiving partial pancreatectomy due to recurrent hypoglycemia 1-2 years after gastric bypass [75].
Table
1.
Evidence of possible neogenesis in the human pancreas |
|
 |
|
Perhaps the most striking evidence of neogenesis in adult humans is a recent study on autopsied pancreas organs from pregnant, post-partum and non-pregnant women [74]. The relative volume of β-cells was increased by up to 40% in the pregnant (n = 18) compared to non-pregnant women (n = 20). There were no differences in replication, apoptosis, or individual cell size, yet the populations of small islets/clusters of β-cells, insulin-positive cells within the ducts, and individual scattered β-cells all doubled. This finding in pregnancy in humans differs from that in mice, in which enhanced replication is the main mechanism [76]. Together, these data strongly support neogenesis as a mechanism for the expansion of β-cells under normal physiological and pathophysiological conditions in humans.
5.2 Pancreas of type 1 diabetes patients
In spite of the commonly held belief that all β-cells are destroyed in type 1 diabetes, a number of reported cases of long-term type 1 diabetes have some islets with a few insulin-positive cells [61, 77-79]. Gianani et al. reported that only 3 of 13 pancreas organs after 10 years duration of childhood onset diabetes had any islets positive for insulin, and 2 of these must be considered as not being classic type 1 patients since there were no insulin-deficient (pseudo-atrophic) islets [80]. In cases studied by Gepts and de Mey, an almost complete loss of β-cells by 1 year was observed if onset was before 7 years of age, yet 40% of 43 patients with onset after 7 years of age and 10-30 years duration of diabetes had some islets with residual β-cells [81] (reviewed in [77]). Similarly, with multiple (8) samples examined per pancreas, Lohr and Kloppel reported 43% of 23 patients with disease duration of between 11 and 54 years had islets with residual β-cells in at least one lobe [61]. Meier et al. reported that 38% of the 42 pancreatic organs from patients with type 1 diabetes and duration ranging from 4 to 67 years had at least some islets with residual β-cells; 88% had scattered β-cells within the parenchyma, but the duration of disease was not provided for the subjects with residual β-cells [78].
In our study of post-mortem pancreata from the well-characterized Joslin 50-year Medalist cohort of people with over 50 years of type 1 diabetes (duration 64.3 ± 1.9 years, n = 28; all but 2 with high risk DR3 and/or DR4 allele), we found that all had scattered single or small clusters of insulin-positive cells in the parenchyma of only some lobes, and 68% had some islets with at least a few insulin-positive cells [82] (and unpublished data). A critical, but unanswerable, question about these pancreata from subjects with long-standing type 1 diabetes is whether the insulin-positive cells have escaped autoimmune destruction or whether they were formed only recently. If the latter were true, these cells could represent the continual attempt at regeneration noted by Warren [2], as quoted at the beginning of this chapter.
6. Conclusions
As outlined above, there is strong evidence that neogenesis happens normally after birth in rodents both in the neonatal period and after some injury, and that it can be induced by different stimuli. It remains the question of the quantitative contribution of postnatal neogenesis to the β-cell mass. Genetically manipulated mice show convincingly that the pathway of neogenesis can be induced in adult rodents under certain conditions. Further work on the stimuli necessary for this induction may provide a path to replenishing β-cells in situ in type 1 diabetes patients.
Disclosure: The authors declare no conflict of interests.
Acknowledgments:
The authors thank those past and present members of the laboratory who have contributed in many ways to exploring the formation of new islet cells and for helpful discussions. The research has been supported by NIDDK, JDRF, and an important group of private donors.
References
- Shapiro AM. Islet transplantation in type 1 diabetes: ongoing challenges, refined procedures, and long-term outcome. Rev Diabet Stud 2012. 9(4):385-406. [DOD] [CrossRef]
- Warren S. The pathology of diabetes mellitus. Second edition, ed. Philadelphia: Lea and Febiger, 1938. [DOD]
- Finegood DT, Scaglia L, Bonner-Weir S. Dynamics of B-cell mass in the growing rat pancreas: estimation with a simple mathematical model. Diabetes 1995. 44:249-256. [DOD] [CrossRef]
- Scaglia L, Cahill CJ, Finegood DT, Bonner-Weir S. Apoptosis participates in the remodeling of the endocrine pancreas in the neonatal rat. Endocrinology 1997. 138(4):1736-1741. [DOD] [CrossRef]
- Bonner-Weir S, Toschi E, Inada A, Reitz P, Fonseca SY, Aye T, Sharma A. The pancreatic ductal epithelium serves as a potential pool of progenitor cells. Pediatric Diabetes 2004. 5:15-22. [DOD] [CrossRef]
- Inada A, Nienaber C, Katsuta H, Fujitani Y, Levine J, Morita R, Sharma A, Bonner-Weir S. Carbonic anhydrase II-positive pancreatic cells are progenitors for both endocrine and exocrine pancreas after birth. Proc Natl Acad Sci U S A 2008. 105:19915-19919. [DOD] [CrossRef]
- Bouwens L, Wang RN, De Blay E, Pipeleers DG, Kloppel G. Cytokeratins as markers of ductal cell differentiation and islet neogenesis in the neonatal rat pancreas. Diabetes 1994. 43:1279-1283. [DOD] [CrossRef]
- Chintinne M, Stange G, Denys B, In't Veld P, Hellemans K, Pipeleers-Marichal M, Ling Z, Pipeleers D. Contribution of postnatally formed small beta cell aggregates to functional beta cell mass in adult rat pancreas. Diabetologia 2010. 53:2380-2388. [DOD] [CrossRef]
- Peng SW, Zhu LY, Chen M, Zhang M, Li DZ, Fu YC, Chen SR, Wei CJ. Heterogeneity in mitotic activity and telomere length implies an important role of young islets in the maintenance of islet mass in the adult pancreas. Endocrinology 2009. 150:3058-3066. [DOD] [CrossRef]
- Seymour PA, Bennett WR, Slack JM. Fission of pancreatic islets during postnatal growth of the mouse. J Anat 2004. 204(2):103-116. [DOD] [CrossRef]
- Jo J, Kilimnik G, Kim A, Guo C, Periwal V, Hara M. Formation of pancreatic islets involves coordinated expansion of small islets and fission of large interconnected islet-like structures. Biophys J 2011. 101:565-574. [DOD] [CrossRef]
- Solar M, Cardalda C, Houbracken I, Martin M, Maestro MA, De Medts N, Xu X, Grau V, Heimberg H, Bouwens L, Ferrer J. Pancreatic exocrine duct cells give rise to insulin-producing beta cells during embryogenesis but not after birth. Dev Cell 2009. 17:849-860. [DOD] [CrossRef]
- Kopinke D, Murtaugh LC. Exocrine-to-endocrine differentiation is detectable only prior to birth in the uninjured mouse pancreas. BMC Dev Biol 2010. 10:38. [DOD] [CrossRef]
- Kopp JL, Dubois CL, Schaffer AE, Hao E, Shih HP, Seymour PA, Ma J, Sander M. Sox9+ ductal cells are multipotent progenitors throughout development but do not produce new endocrine cells in the normal or injured adult pancreas. Development 2011. 138:653-665. [DOD] [CrossRef]
- Furuyama K, Kawaguchi Y, Akiyama H, Horiguchi M, Kodama S, Kuhara T, Hosokawa S, Elbahrawy A, Soeda T, Koizumi M, et al. Continuous cell supply from a Sox9-expressing progenitor zone in adult liver, exocrine pancreas and intestine. Nat Genet 2011. 43:34-41. [DOD] [CrossRef]
- Inada A, Nienaber C, Fonseca S, Bonner-Weir S. Timing and expression pattern of carbonic anhydrase II in pancreas. Dev Dyn 2006. 235:1571-1577. [DOD] [CrossRef]
- Ouziel-Yahalom LW, Straussman S, Guo L, Sharma A, Weir GC, Bonner-Weir S. Ductal heterogeneity suggests a subpopulation serves as postnatal pancreatic progenitors. Diabetes 2010. 59(Suppl 1):A25. [DOD]
- Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 2004. 429:41-46. [DOD] [CrossRef]
- Guo C, Yang W, Lobe CG. A Cre recombinase transgene with mosaic, widespread tamoxifen-inducible action. Genesis 2002. 32:8-18. [DOD] [CrossRef]
- Reinert RB, Kantz J, Misfeldt AA, Poffenberger G, Gannon M, Brissova M, Powers AC. Tamoxifen-Induced Cre-loxP Recombination Is Prolonged in Pancreatic Islets of Adult Mice. Plos One 2012. 7:e33529. [DOD] [CrossRef]
- Nakamura K, Minami K, Tamura K, Iemoto K, Miki T, Seino S. Pancreatic beta-cells are generated by neogenesis from non-beta-cells after birth. Biomed Res 2011. 32:167-174. [DOD] [CrossRef]
- Bonner-Weir S, Baxter LA, Schuppin GT, Smith FE. A second pathway for regeneration of the adult exocrine and endocrine pancreas: A possible recapitulation of embryonic development. Diabetes 1993. 42:1715-1720. [DOD] [CrossRef]
- Wang RN, Kloppel G, Bouwens L. Duct- to islet-cell differentiation and islet growth in the pancreas of duct-ligated adult rats. Diabetologia 1995. 38:1405-1411. [DOD] [CrossRef]
- Weaver CV, Sorenson RL, Kaung HC. Immunocytochemical localization of insulin-immunoreactive cells in the ducts of rats treated with trypsin inhibitor. Diabetologia 1985. 28:781-785. [DOD]
- Xu G, Stoffers DA, Habener JF, Bonner-Weir S. Exendin-4 stimulates both beta-cell replication and neogenesis, resulting in increased beta-cell mass and improved glucose tolerance in diabetic rats. Diabetes 1999. 48:2270-2276. [DOD] [CrossRef]
- Waguri M, Yamamoto K, Miyagawa J, Tochino Y, Yamamori K, Kajimoto Y, Nakajima H, Watada H, Yoshiuchi I, Itoh N, et al. Demonstration of two different processes of B-cell regeneration in a new diabetic mouse model induced by selective perfusion of alloxan. Diabetes 1997. 46:1281-1290. [DOD] [CrossRef]
- Rosenberg L, Duguid WP, Vinik AI. The effect of cellophane wrapping of the pancreas in the Syrian golden hamster: autoradiographic observations. Pancreas 1989. 4:31-37. [DOD] [CrossRef]
- Gu D, Sarvetnick N. Epithelial cell proliferation and islet neogenesis in IFN-gamma transgenic mice. Development 1993. 118:33-46. [DOD]
- Wang TC, Bonner-Weir S, Oates PS, Chulak M, Simon B, Merlino GT, Schmidt EV, Brand SJ. Pancreatic gastrin stimulates islet differentiation of transforming growth factor alpha-induced ductular precursor cells. J Clin Invest 1993. 92:1349-1356. [DOD] [CrossRef]
- Collombat P, Xu X, Ravassard P, Sosa-Pineda B, Dussaud S, Billestrup N, Madsen OD, Serup P, Heimberg H, Mansouri A. The ectopic expression of Pax4 in the mouse pancreas converts progenitor cells into alpha and subsequently beta cells. Cell 2009. 138:449-462. [DOD] [CrossRef]
- Bonner-Weir S, Trent DF, Weir GC. Partial pancreatectomy in the rat and subsequent defect in glucose-induced insulin release. J Clin Invest 1983. 71:1544-1553. [DOD] [CrossRef]
- Brockenbrough JS, Weir GC, Bonner-Weir S. Discordance of exocrine and endocrine growth after 90% pancreatectomy in rats. Diabetes 1988. 37:232-236. [DOD] [CrossRef]
- Jetton TL, Lausier J, LaRock K, Trotman WE, Larmie B, Habibovic A, Peshavaria M, Leahy JL. Mechanisms of compensatory beta-cell growth in insulin-resistant rats: roles of Akt kinase. Diabetes 2005. 54:2294-2304. [DOD] [CrossRef]
- Bonner-Weir S, Stubbs M, Reitz P, Taneja M, Smith FE. Partial pancreatectomy as a model of pancreatic regeneration. In: Sarvetnick N (ed). Pancreas regeneration. Karger Landes Systems, 1997, 138-153. [DOD]
- Peshavaria M, Larmie BL, Lausier J, Satish B, Habibovic A, Roskens V, Larock K, Everill B, Leahy JL, Jetton TL. Regulation of pancreatic beta-cell regeneration in the normoglycemic 60% partial-pancreatectomy mouse. Diabetes 2006. 55:3289-3298. [DOD] [CrossRef]
- Ackermann Misfeldt A, Costa RH, Gannon M. Beta-cell proliferation, but not neogenesis, following 60% partial pancreatectomy is impaired in the absence of FoxM1. Diabetes 2008. 57:3069-3077. [DOD] [CrossRef]
- Teta M, Rankin MM, Long SY, Stein GM, Kushner JA. Growth and regeneration of adult beta cells does not involve specialized progenitors. Dev Cell 2007. 12:817-826. [DOD] [CrossRef]
- Lee CS, De Leon DD, Kaestner KH, Stoffers DA. Regeneration of pancreatic islets after partial pancreatectomy in mice does not involve the reactivation of neurogenin-3. Diabetes 2006. 55:269-272. [DOD]
- Bonner-Weir S, Leahy JL, Weir GC. Induced rat models of non-insulin-dependent diabetes mellitus. In: Shapiro E, Renold AE (eds). Lessons from animal diabetes II. John Libbey, London, 1988, 295-300. [DOD]
- Li WC, Rukstalis JM, Nishimura W, Tchipashvili V, Habener JF, Sharma A, Bonner-Weir S. Activation of pancreatic duct-derived progenitor cells during pancreatic regeneration in adult rats. J Cell Science 2010. 123(Pt 16):2792-2802. [DOD]
- Sharma A, Zangen GH, Reitz P, Taneja M, Lissauer ME, Miller CP, Weir GC, Habener JF, Bonner-Weir S. The homeodomain protein IDX-1 increases after an early burst of proliferation during pancreatic regeneration. Diabetes 1999. 48:507-513. [DOD] [CrossRef]
- Hultquist GT, Karlsson U, Hallner AC. The regenerative capacity of the pancreas in duct-ligated rats. Exp Pathol 1979. 17:44-52. [DOD]
- Edstrom C. Further quantitative structural studies of the pancreatic islet parenchyma in rats with duct ligation. Acta Soc Med Uppsala 1971. 76:127-138. [DOD]
- Lardon J, Huyens N, Rooman I, Bouwens L. Exocrine cell transdifferentiation in dexamethasone-treated rat pancreas. Virchows Arch 2004. 444:61-65. [DOD] [CrossRef]
- Rooman I, Lardon J, Bouwens L. Gastrin stimulates beta-cell neogenesis and increases islet mass from transdifferentiated but not from normal exocrine pancreas tissue. Diabetes 2002. 51:686-690. [DOD] [CrossRef]
- Xu X, D'Hoker J, Stange G, Bonne S, De Leu N, Xiao X, Van De Casteele M, Mellitzer G, Ling Z, Pipeleers D, Bouwens L, Scharfmann R, Gradwohl G, Heimberg H. Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell 2008. 132:197-207. [DOD] [CrossRef]
- Chintinne M, Stange G, Denys B, Ling Z, In't Veld P, Pipeleers D. Beta cell count instead of beta cell mass to assess and localize growth in beta cell population following pancreatic duct ligation in mice. Plos One 2012. 7:e43959. [DOD] [CrossRef]
- Criscimanna A, Speicher JA, Houshmand G, Shiota C, Prasadan K, Ji B, Logsdon CD, Gittes GK, Esni F. Duct cells contribute to regeneration of endocrine and acinar cells following pancreatic damage in adult mice. Gastroenterology 2011. 141:1451-1462. [DOD] [CrossRef]
- Sarvetnick NE, Gu D. Regeneration of pancreatic endocrine cells in interferon-gamma transgenic mice. Adv Exp Med Biol 1992. 321:85-89. [DOD] [CrossRef]
- Kritzik MR, Krahl T, Good A, Krakowski M, St-Onge L, Gruss P, Wright C, Sarvetnick N. Transcription factor expression during pancreatic islet regeneration. Mol Cell Endocrinol 2000. 164:99-107. [DOD] [CrossRef]
- Kritzik MR, Jones E, Chen Z, Krakowski M, Krahl T, Good A, Wright C, Fox H, Sarvetnick N. PDX-1 and Msx-2 expression in the regenerating and developing pancreas. J Endocrinol 1999. 163:523-530. [DOD] [CrossRef]
- Song SY, Gannon M, Washington MK, Scoggins CR, Meszoely IM, Goldenring JR, Marino CR, Sandgren EP, Coffey RJ Jr, Wright CV, Leach SD. Expansion of Pdx1-expressing pancreatic epithelium and islet neogenesis in transgenic mice overexpressing transforming growth factor alpha. Gastroenterology 1999. 117(6):1416-1426. [DOD] [CrossRef]
- Jhappan C, Stahle C, Harkins RN, Fausto N, Smith GH, Merlino GT. TGFalpha overexpression in transgenic mice induces liver neoplasia and abnormal development of the mammary gland and pancreas. Cell 1990. 61:1137-1146. [DOD] [CrossRef]
- Zhang H, Zhang J, Pope CF, Crawford LA, Vasavada RC, Jagasia SM, Gannon M. Gestational diabetes mellitus resulting from impaired beta-cell compensation in the absence of FoxM1, a novel downstream effector of placental lactogen. Diabetes 2009. 59:143-152. [DOD] [CrossRef]
- Aye T, Toschi E, Sharma A, Sgroi D, Bonner-Weir S. Identification of markers for newly formed beta-cells in the perinatal period: a time of recognized beta-cell immaturity. J Histochem Cytochem 2010. 58:369-376. [DOD] [CrossRef]
- Toschi E AT, Fuller A, Gaudet J, Sharma A, Weir G. C, Sgroi D, Bonner-Weir S. Gene expression profiling of new beta cells: evidence for differentiating beta cells passing through a ductal phenotype. Diabetes 2004. 53(Suppl 2):1613. [DOD]
- Yatoh S, Dodge R, Akashi T, Omer A, Sharma A, Weir GC, Bonner-Weir S. Differentiation of affinity-purified human pancreatic duct cells to beta-cells. Diabetes 2007. 56:1802-1809. [DOD] [CrossRef]
- Meier JJ, Butler AE, Saisho Y, Monchamp T, Galasso R, Bhushan A, Rizza RA, Butler PC. Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes 2008. 57:1584-1594. [DOD] [CrossRef]
- Saisho Y, Butler AE, Meier JJ, Monchamp T, Allen-Auerbach M, Rizza RA, Butler PC. Pancreas volumes in humans from birth to age one hundred taking into account sex, obesity, and presence of type-2 diabetes. Clin Anat 2007. 20:933-942. [DOD] [CrossRef]
- Gregg BE, Moore PC, Demozay D, Hall BA, Li M, Husain A, Wright AJ, Atkinson MA, Rhodes CJ. Formation of a human beta-cell population within pancreatic islets is set early in life. J Clin Endocrinol Metab 2012. 97:3197-3206. [DOD] [CrossRef]
- Lohr M, Kloppel G. Residual insulin positivity and pancreatic atrophy in relation to duration of chronic type 1 (insulin-dependent) diabetes mellitus and microangiopathy. Diabetologia 1987. 30:757-762. [DOD] [CrossRef]
- Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003. 52:102-110. [DOD] [CrossRef]
- Yoon KH, Ko SH, Cho JH, Lee JM, Ahn YB, Song KH, Yoo SJ, Kang MI, Cha BY, Lee KW, Son HY, Kang SK, Kim DG, Lee IK, Bonner-Weir S. Selective beta-cell loss and alpha-cell expansion in patients with type 2 diabetes mellitus in Korea. J Clin Endoc Metab 2003. 88:2300-2308. [DOD] [CrossRef]
- Rahier J, Guiot Y, Goebbels RM, Sempoux C, Henquin JC. Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes Metab 2008. 10(Suppl 4):32-42. [DOD] [CrossRef]
- Hanley SC, Austin E, Assouline-Thomas B, Kapeluto J, Blaichman J, Moosavi M, Petropavlovskaia M, Rosenberg L. Beta-cell mass dynamics and islet cell plasticity in human type 2 diabetes. Endocrinology 2010. 151:1462-1472. [DOD] [CrossRef]
- Kloppel G, Lohr M, Habich K, Oberholzer M, Heitz PU. Islet pathology and the pathogenesis of type 1 and type 2 diabetes mellitus revisited. Surv Synth Pathol Res 1985. 4:110-125. [DOD]
- Tyrberg B, Ustinov J, Otonkoski T, Andersson A. Stimulated endocrine cell proliferation and differentiation in transplanted human pancreatic islets: effects of the ob gene and compensatory growth of the implantation organ. Diabetes 2001. 50:301-307. [DOD] [CrossRef]
- Alarcon C, Leahy JL, Schuppin GT, Rhodes CJ. Increased secretory demand rather than a defect in the proinsulin conversion mechanism causes hyperproinsulinemia in a glucose-infusion rat model of non-insulin-dependent diabetes mellitus. J Clin Invest 1995. 95:1032-1039. [DOD] [CrossRef]
- Reers C, Erbel S, Esposito I, Schmied B, Buchler MW, Nawroth PP, Ritzel RA. Impaired islet turnover in human donor pancreata with aging. Eur J Endocrinol 2009. 160:185-191. [DOD] [CrossRef]
- Caballero F, Siniakowicz S, Hollister-Lock J, Duran L, Katsuta H, Yamada T, Lei J, Deng S, Westermark GT, Markmann J, Bonner-Weir S, Weir GC. Human beta-cell birth and death in pancreas from cadaver donors, autopsies, surgical specimens, and islets transplanted into mice. Cell Transplant 2013. In press. [DOD]
- Meier JJ, Butler AE, Galasso R, Butler PC. Hyperinsulinemic hypoglycemia after gastric bypass surgery is not accompanied by islet hyperplasia or increased beta-cell turnover. Diabetes Care 2006. 29:1554-1559. [DOD] [CrossRef]
- Meier JJ, Butler AE, Galasso R, Rizza RA, Butler PC. Increased islet beta cell replication adjacent to intrapancreatic gastrinomas in humans. Diabetologia 2006. 49:2689-2696. [DOD] [CrossRef]
- Phillips JM, O'Reilly L, Bland C, Foulis AK, Cooke A. Patients with chronic pancreatitis have islet progenitor cells in their ducts, but reversal of overt diabetes in NOD mice by anti-CD3 shows no evidence for islet regeneration. Diabetes 2007. 56:634-640. [DOD] [CrossRef]
- Butler AE, Cao-Minh L, Galasso R, Rizza RA, Corradin A, Cobelli C, Butler PC. Adaptive changes in pancreatic beta cell fractional area and beta cell turnover in human pregnancy. Diabetologia 2010. 53(10):2167-2176. [DOD] [CrossRef]
- Patti ME, McMahon G, Mun EC, Bitton A, Holst JJ, Goldsmith J, Hanto DW, Callery M, Arky R, Nose V, Bonner-Weir S, Goldfine AB. Severe hypoglycaemia post-gastric bypass requiring partial pancreatectomy: evidence for inappropriate insulin secretion and pancreatic islet hyperplasia. Diabetologia 2005. 48:2236-2240. [DOD] [CrossRef]
- Parsons JA, Bartke A, Sorenson RL. Number and size of islets of langerhans in pregnant human growth hormone-expressing transgenic, and pituitary dwarf mice: effect of lactogenic hormones. Endocrinology 1995. 136:2013-2021. [DOD] [CrossRef]
- Pipeleers D, Ling Z. Pancreatic beta cells in insulin-dependent diabetes. Diabetes Metab Rev 1992. 8:209-227. [DOD] [CrossRef]
- Meier JJ, Bhushan A, Butler AE, Rizza RA, Butler PC. Sustained beta cell apoptosis in patients with long-standing type 1 diabetes: indirect evidence for islet regeneration? Diabetologia 2005. 48:2221-2228. [DOD]
- Stefan Y, Orci L, Malaisse-Lagae F, Perrelet A, Patel Y, Unger RH. Quantitation of endocrine cell content in the pancreas of nondiabetic and diabetic humans. Diabetes 1982. 31:694-700. [DOD] [CrossRef]
- Gianani R, Campbell-Thompson M, Sarkar SA, Wasserfall C, Pugliese A, Solis JM, Kent SC, Hering BJ, West E, Steck A, et al. Dimorphic histopathology of long-standing childhood-onset diabetes. Diabetologia 2010. 53:690-698. [DOD] [CrossRef]
- Gepts W, De Mey J. Islet cell survival determined by morphology. An immunocytochemical study of the islets of Langerhans in juvenile diabetes mellitus. Diabetes 1978. 27(Suppl 1):251-261. [DOD]
- Keenan HA, Sun JK, Levine J, Doria A, Aiello LP, Eisenbarth G, Bonner-Weir S, King GL. Residual insulin production and pancreatic ss-cell turnover after 50 years of diabetes: Joslin Medalist Study. Diabetes 2010. 59:2846-2853. [DOD] [CrossRef]
- Meier JJ, Lin JC, Butler AE, Galasso R, Martinez DS, Butler PC. Direct evidence of attempted beta cell regeneration in an 89-year-old patient with recent-onset type 1 diabetes. Diabetologia 2006. 49:1838-1844. [DOD] [CrossRef]
- Martin-Pagola A, Sisino G, Allende G, Dominguez-Bendala J, Gianani R, Reijonen H, Nepom GT, Ricordi C, Ruiz P, Sageshima J, Ciancio G, Burke GW, Pugliese A. Insulin protein and proliferation in ductal cells in the transplanted pancreas of patients with type 1 diabetes and recurrence of autoimmunity. Diabetologia 2008. 51:1803-1813. [DOD] [CrossRef]
This article has been cited by other articles:

|
Pancreatic beta-cell regeneration: Facultative or dedicated progenitors?
Afelik S, Rovira M
Mol Cell Endocrinol 2017. 445:85-94
|
|
|
Bariatric surgery influences beta-cell turnover in non obese rats
Camacho-Ramirez A, Blandino-Rosano M, Segundo-Iglesias MC, Lechuga-Sancho AM, Aguilar-Diosdado M, Perez-Arana GM, Prada-Oliveira JA
Histol Histopathol 2017. In press
|
|

|
Stress-induced adaptive islet cell identity changes
Cigliola V, Thorel F, Chera S, Herrera PL
Diabetes Obes Metab 2016. 18(Suppl 1):87-96
|
|

|
New Insights into Diabetes Cell Therapy
Lysy PA, Corritore E, Sokal EM
Curr Diab Rep 2016. 16(5):38
|
|

|
Unique Aspects of Cryptochrome in Chronobiology and Metabolism, Pancreatic Beta-Cell Dysfunction, and Regeneration: Research into Cysteine414-Alanine Mutant CRY1
Okano S
J Diabetes Res 2016. 2016:3459246
|
|

|
Beta-cell replacement sources for type 1 diabetes: a focus on pancreatic ductal cells
Corritore E, Lee YS, Sokal EM, Lysy PA
Ther Adv Endocrinol Metab 2016. 7(4):182-199
|
|

|
Beta-Cell dedifferentiation, reduced duct cell plasticity, and impaired β-cell mass regeneration in middle-aged rats
Tellez N, Vilaseca M, Marti Y, Pla A, Montanya E
Am J Physiol Endocrinol Metab 2016. 311(3):E554-E563
|
|

|
The Human Endocrine Pancreas: New Insights on Replacement and Regeneration
Dominguez-Bendala J, Lanzoni G, Klein D, Alvarez-Cubela S, Pastori RL
Trends Endocrinol Metab 2016. 27(3):153-162
|
|

|
The Rise and the Fall of Betatrophin/ANGPTL8 as an Inducer of beta-Cell Proliferation
Abu-Farha M, Al Madhoun A, Abubaker J
J Diabetes Res 2016. 2016:4860595
|
|
|
Cellular therapy of diabetes: focus on the latest developments
Lysy PA
Med Sci (Paris) 2016. 32(4):401-407
|
|

|
Antidiabetic and Antioxidant Impacts of Desert Date (Balanites aegyptiaca) and Parsley (Petroselinum sativum) Aqueous Extracts: Lessons from Experimental Rats
Abou Khalil NS, Abou-Elhamd AS, Wasfy SI, El Mileegy IM, Hamed MY, Ageely HM
J Diabetes Res 2016. 2016:8408326
|
|

|
Beta-Cell Identity in Type 2 Diabetes: Lost or Found?
Butler AE, Dhawan S
Diabetes 2015. 64(8):2698-2700
|
|

|
The double trouble of metabolic diseases: the diabetes-cancer link
Tudzarova S, Osman MA
Mol Biol Cell 2015. 26(18):3129-3139
|
|

|
Development, growth and maintenance of β-cell mass: models are also part of the story
Khadra A, Schnell S
Mol Aspects Med 2015. 42:78-90
|
|

|
Direct Reprogramming for Pancreatic Beta-Cells Using Key Developmental Genes
Cavelti-Weder C, Li W, Zumsteg A, Stemann M, Yamada T, Bonner-Weir S, Weir G, Zhou Q
Curr Pathobiol Rep 2015. 3(1):57-65
|
|

|
Beta-cell induction in vivo in severely diabetic male mice by changing the circulating levels and pattern of the ratios of estradiol to androgens
Inada A, Inada O, Fujii NL, Fujishima K, Inai T, Fujii H, Sueishi K, Kurachi K
Endocrinology 2014. 155(10):3829-3842
|
|

|
Maturation of stem cell-derived beta-cells guided by the expression of urocortin 3
van der Meulen T, Huising MO
Rev Diabet Stud 2014. 11(1):115-132
|
|

|
In vitro differentiation and expansion of human pluripotent stem cell-derived pancreatic progenitors
Chmielowiec J, Borowiak M
Rev Diabet Stud 2014. 11(1):19-34
|
|
|


)
)

