Chapter IV. Clinical Trials
| Rev Diabet Stud,
2012,
9(4):372-381 |
DOI 10.1900/RDS.2012.9.372 |
CD3 Monoclonal Antibodies: A First Step Towards Operational Immune Tolerance in the Clinic
Lucienne Chatenoud1,2, Herman Waldmann3
1INSERM U1013, Necker Hospital, Paris, France
2Universite Paris Descartes, Sorbonne Paris Cite, Paris, France
3University of Oxford, Sir William Dunn School of Pathology, South Parks Road, Oxford OX1 3RE, UK
Address correspondence to: Lucienne Chatenoud, e-mail lucienne.chatenoud@inserm.fr, or Herman Waldmann, e-mail herman.waldmann@path.ox.ac.uk
Manuscript submitted December 19, 2012; resubmitted January 31, 2013; accepted February 9, 2013.
Keywords: type 1 diabetes, immune tolerance, anti-CD3 antibody, OKT3, beta-cell, Fc receptor
Abstract
Type 1 diabetes (T1D) is a prototypic organ-specific autoimmune disease resulting from the selective destruction of insulin-secreting β-cells within the pancreatic islets of Langerhans. It is caused by an immune-mediated inflammation, involving autoreactive CD4+ and CD8+ T lymphocytes that infiltrate the islets and initiate insulitis. The use of exogenous insulin is the current standard treatment. However, in spite of significant advances, this therapy is still associated with major constraints, including risk of hypoglycemia and severe degenerative complications. As T1D mainly affects children and young adults, any candidate immune therapy must be safe, and it must avoid a sustained depression of immune responses with all its attendant problems of recurrent infection and drug toxicity. In this context, inducing or restoring immune tolerance to target autoantigens would be the ideal approach. We refer to immune tolerance here as the selective damping of the damaging autoimmune response following a short treatment, while keeping intact the capacity of the host to respond normally to exogenous antigens. The therapeutic approach we discuss in this article originates from attempts to induce tolerance both to soluble antigens and tissue antigens (i.e. alloantigens and autoantigens) by using biological agents that selectively interfere with lymphocyte activation, namely polyclonal and monoclonal anti-T cell antibodies. The challenged dogma was that, in an adult-primed immune system, it was not possible to restore self-tolerance therapeutically without the use of exogenous autoantigen administration. The reality has been that, in diabetes, endogenous host autoantigen can fulfill this role because a significant amount of functioning β-cells remains, even at the time of established hyperglycemia. Experimental results obtained in the 1990s showed that a short-term CD3 antibody treatment in recently diagnosed diabetic non-obese diabetic (NOD) mice induced permanent remission of the disease by restoring self-tolerance. Based on these findings, phase I, II, and III trials were conducted using two distinct humanized Fc-mutated antibodies to human CD3, namely ChAglyCD3 (otelixizumab) and OKT3γ1 Ala-Ala (teplizumab). Overall, when dosing was adequate, the results demonstrated that CD3 antibodies preserved β-cell function very efficiently, maintaining significantly high levels of endogenous insulin secretion in treated patients for up to 24 months after treatment. These data provided the first proof of concept for a long-term therapeutic effect in T1D following a short course administration of a therapeutic agent. Our aim is to review these data and to discuss them in the context of the pitfalls linked to pharmaceutical development, especially in the context of pediatric patients, as in autoimmune diabetes.
Abbreviations: ADCC – antibody-dependent cell-mediated cytotoxicity; APC - antigen-presenting cell; CDR – complementarity-determining region; EAE - experimental allergic encephalomyelitis; EBV – Epstein Barr virus; F(ab) – fragment antigen binding; Fc – fragment, crystalizable; HbA1c – glycosylated hemoglobin; IFNγ – interferon gamma; Ig – immunoglobulin; IGRP – islet-specific glucose-6-phosphatase catalytic subunit related protein; IL – interleukin; iTreg – induced Treg; MOG – myelin oligodendrocyte glycoprotein; mRNA – messenger RNA; NOD – non-obese diabetic; OKT3 – ortho Kung T cell 3; PCR – polymerase chain reaction; PLP – proteolipid protein; RNA – ribonucleic acid; SJL – Swiss/Jackson Laboratory; T1D – type 1 diabetes; TCR – T cell receptor; TGFβ – transforming growth factor beta; Th – T helper; TNF – tumor necrosis factor; TNP-KLH – 2,4,6-trinitrophenol-conjugated keyhole limpet hemocyanin; Treg – regulatory T cell
1. Introduction
Converging evidence from animal models and clinical trials has demonstrated that a key component of the pathogenesis of type 1 diabetes (T1D) is the autoimmune reaction to β-cell autoantigens and the associated inflammation [1-3]. Although a triggering role of certain environmental factors (e.g. viruses) and a genetically determined susceptibility of β-cells to such factors must not be disregarded, the initiation and extent of the subsequent β-cell destruction are due to an interplay between innate and adaptive immune responses. This concept forms the basis for efforts to counteract the immune attack in order to durably arrest T1D, as chronic administration of insulin is only a substitutive treatment that does not prevent the development of chronic degenerative complications. Importantly, current epidemiological studies predict a dramatic impact of T1D on public health in the near future [4]. The disease incidence will continue to significantly increase in the coming decade, and the pathology will proportionally affect mainly very young children under 5 years of age [4].
Any T1D immune-therapeutic intervention must build on known approaches for manipulating autoimmune mechanisms to devise novel therapeutic strategies that address this burgeoning unmet medical need [4]. In the context of the young patient population increasingly affected by T1D, the challenge is to obtain clinical efficacy without compromising the host defense against infections and tumors. This is the rationale to re-establish immune tolerance to β-cell autoantigens.
The autoimmune reaction develops in three major stages, as outlined below; each of which is a potential therapeutic target [5]:
1. The first stage is the recognition of β-cell autoantigens by T cells, with T cell activation triggered by autoantigens. It is not sufficient that T cells merely see the antigen, but recognition must be associated with the expression of a series of molecules expressed on the membrane of antigen-presenting cells (APCs), in particular dendritic cells, in a well-defined cellular activation context.
2. The second stage constitutes the maturation of autoreactive T cells, i.e. the proliferation and differentiation of T cells into pathogenic effectors, progressively leading to their accumulation in the islets of Langerhans. When their numbers and functional (i.e. damaging) capacity reaches or exceeds a certain threshold, they begin to attack β-cell in an interplay with the innate immune system. A key point is that this autoimmune and inflammatory response, like so many other immune reactions, is under the control of a CD4+ population of regulatory T cells (Tregs), including both natural Tregs [6-7], a lineage directly emerging from the thymus, and induced Tregs (iTregs) that originate from mature precursors in the periphery [8-9].
3. The third stage is the β-cell attack, leading to their destruction by apoptosis [10-11]. However, β-cell death appears to be preceded by a relatively long intermediate period of partial or complete functional inhibition which, importantly, may be reversible [12-13]. β-cells do not behave like inert targets; the efficiency of the autoimmune attack is influenced by the inherent differentiation-dependent β-cell ‘sensitivity’ to the aggression [3]. Hence the importance of addressing mechanisms regarding β-cell resistance to damage.
A promising immune intervention strategy is the use of monoclonal antibodies targeting CD3, a molecule associated with the T cell receptor (TCR) for antigen recognition [14]. CD3 monoclonal antibodies induce partial T cell depletion and interfere with T cell activation. In the 1990s, it was established in non-obese diabetic (NOD) mice that a short low-dose treatment with a CD3 antibody induced durable remission of established T1D by restoring immune tolerance [15]. In 2000, when humanized "disabled" non-Fc receptor-binding anti-human CD3 antibodies became available, we conducted a phase II placebo-controlled trial that successfully translated this preclinical finding to the clinic in recent onset T1D. Thus, a single one week treatment course, in patients with recent-onset T1D, induced a significant preservation of C-peptide and a decrease in insulin requirements for over 2-3 years [16-17]. These pivotal results fostered the pharmaceutical development of CD3 antibodies, and phase III trials were launched. Our aim here is to present and discuss the data that have validated CD3 antibodies as a target class, with emphasis on patient variability regarding preservation of β-cell function, and the need to carefully consider safety issues.
2. Clinical development of CD3 antibodies: a story spanning more than 20 years
2.1 The first experiences in clinical transplantation
The history of the discovery and clinical application of CD3 antibodies was an amazing sequence of events, marked by a major scientific discovery, much empiricism, and a good dose of chance (Table 1). The major scientific discovery was the development of a technique to produce murine hybridoma-secreting monoclonal antibodies, reported by H. Kohler and C. Milstein in the 1970s, which earned them the Nobel prize [18]. Within 4 years, P. Kung and G. Goldstein, working at Ortho (Raritan, USA), published the first three mouse monoclonal antibodies to human T cell surface antigens, including OKT3 ('O' standing for Ortho, 'K' for Kung, 'T' for T cell and '3' the ranking order in the lab book) [19]. Given its ability to recognize all mature human T cells and to block the in vitro response of lymphocytes to alloantigens (i.e. blockade of both the proliferation and the generation of cytotoxic effectors), OKT3 had much in common with anti-lymphocyte polyclonal sera, which were already used in clinical organ transplantation at that time. Based on these practical, yet pretty sketchy, facts, and while its precise molecular target was still unknown, OKT3 (muromomab) was the first monoclonal antibody to enter clinical practice for transplantation in 1981, the first to treat acute renal allograft rejection episodes and then to prevent rejection [20-22].
Table
1.
Milestones in the clinical development of CD3 monoclonal antibodies |
|
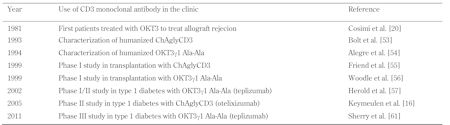 |
 |
A few years after the first patient was treated, the molecular target of OKT3 proved to be nothing less than one of the chains (ε) of the CD3 molecular complex, recognized as the transducing element of the TCR for antigen recognition [14, 23]. Quite paradoxically, in these early days, OKT3 benefited from an impressively rapid development due to its very potent immunosuppressive activity, and was licensed by 1984 with absolutely no preclinical data available. In fact, as with most anti-T cell monoclonal antibodies, CD3 antibodies are highly "species-specific". Human CD3 antibodies do not cross-react with lymphocytes from other animate beings, not even with those from common non-human primates such as Rhesus or Cynomolgus; they only recognize chimpanzee cells [24]. This explains why OKT3 was not subjected to conventional toxicological analysis, which would have revealed its Fc receptor dependency and the in vivo counterpart of its well documented in vitro T cell mitogenicity [25], namely its characteristic cytokine release-mediated 'flu-like' syndrome. Thus, following the first OKT3 injection, a massive yet self-limited systemic release of various cytokines, mostly T cell derived, including TNF, IL-6, IFNγ, and IL-10 was observed [26-28]. Needless to say, the risk would have been high for this drug to be discarded from further development if conventional toxicology had been performed. Because of this side effect, and the fact that better tolerated biological immunosuppressants subsequently emerged, OKT3 was completely abandoned.
2.2 CD3 antibodies as mediators of immune tolerance in primed hosts: the proof-of-concept in experimental autoimmunity
The characterization of CD3 antibodies reacting to mouse and rat T cells [29-31] fostered the development of experimental models, which rapidly demonstrated that CD3 antibodies are able to promote immune tolerance in organ allograft transplantations and autoimmunity far beyond their capacity to mediate a potent immunosuppressive activity [30, 32-33]. In a rat transplant model, B, Hall et al. were the first to report that a non-mitogenic CD3-specific antibody administered over a short course of time, induced permanent survival of fully mismatched vascularized heart grafts. Secondary donor-matched skin grafts survived indefinitely, while third-party skin allografts were normally rejected [30, 32-33]. Early post-transplant, CD3-specific antibody-treated recipients showed a typical Th2 immune deviation as reflected by increased T cell IL-4 and IL-5 mRNA levels [32]. Hayward and Schreiber reported that a single CD3 antibody injection in newborn NOD mice almost completely prevented the occurrence of diabetes, i.e. at 8 months of age only 10% of the treated NOD mice had diabetes as compared to 90-95% in untreated controls [33]. These results clearly illustrated the capacity of CD3 antibodies to induce immune tolerance to β-cell antigens when targeting the neonatal repertoire.
More interestingly and quite unexpectedly, we first reported that, when applied to overtly diabetic NOD mice, CD3 antibodies expressed the unique ability to restore self-tolerance once target destruction was ongoing and advanced, a capacity that clearly distinguished them from other tolerance-promoting immune therapy strategies in diabetes (i.e. delivery of β-cell autoantigens) [15, 34-36]. Thus, a short five-day low-dose treatment with CD3-specific antibodies, at the time of established hyperglycemia, induced long-lasting disease remission in the absence of any insulin treatment. [15, 35-36]. This effect resulted from restoration of immune tolerance to islet antigens since syngeneic islet grafts survived indefinitely in mice showing CD3-specific antibody-induced remission, while the grafts were rapidly destroyed by autoimmunity when implanted into untreated diabetic NOD females. In contrast, skin allografts were rejected as usual in CD3-specific antibody-treated NOD mice [15].
Similar results were reported in experimental allergic encephalomyelitis (EAE) induced by proteolipid protein (PLP) in lean SJL mice [37]. The treatment was highly effective by affecting ongoing PLP-specific biological and clinical responses. In contrast, no effect at all was observed for disease prevention when the antibody was given concomitantly with PLP administration. Most remarkably, the treatment was still active at the peak of the disease, which accelerated disease remission and prevented further relapses [37]. Comparable results were reported in myelin oligodendrocyte glycoprotein (MOG)-induced EAE [38]. It is also of some interest that oral administration of CD3 antibody could both prevent and treat PLP-induced EAE in SJL mice and MOG-induced EAE in NOD mice [39].
Last, but not least, in a mouse model of induced Th1-mediated inflammatory bowel disease [40], in IL-2 deficient (IL2-/-) mice following administration of TNP-KLH (2,4,6-trinitrophenol-conjugated keyhole limpet hemocyanin), administration of a CD3 antibody inhibited colitis, an effect associated with blockade of IFNγ production and recovery of TGFβ secretion by lamina propria T cells.
2.3 Mechanistic considerations
CD3 antibody administration induces partial T cell depletion that affects about 50% of the host’s CD3/TCR+ cells when Fc receptor-binding, mitogenic CD3 antibodies are used (i.e. the hamster 145 2C11 antibody), and 20-30% with non-mitogenic antibodies (in mice, this is especially the case when F(ab)'2 fragments of 145 2C11 are used) [35-36, 41]. To assess such depletion objectively, it has been important to analyze the spleen and lymph nodes of treated mice. In patients, the only compartment accessible is the peripheral blood, and the data obtained from blood samples are biased by cytokine release, even if minimal. Thus, the standard picture observed with OKT3 was defined as the disappearance of all T cells from the circulation 30-60 minutes after the first injection [42]. This is caused by cytokine-release that favors the activation of endothelial cells, leading to increased adhesiveness, lymphocyte elimination [43], and cell redistribution within the various compartments, ultimately with engulfment by reticuloendothelial cells. With the "disabled" CD3 antibodies, any antibody-induced depletion does not depend on complement fixation or antibody-dependent cell-mediated cytotoxicity (ADCC). Rather, it depends on the antibody's fine specificity that causes redirection of T cell lysis by bridging cytotoxic T cells to other T cells [44] and the induction of apoptosis or programmed cell death (to which activated cells appear particularly sensitive) [45]. Recent data show that antigen-activated T cells appear especially sensitive to the CD3 antibody-mediated effect, while Tregs are more resistant [46-48].
T cells, which are not eliminated during CD3 antibody treatment, undergo antigenic modulation of CD3/TCR. Specifically, the receptor complex transiently disappears upon antibody binding, following capping and internalization or shedding. These cells stain as CD3-TCR-CD4+ or CD3-TCR-CD8+ [42, 49]. CD3 antibody-modulated cells are, of course, unresponsive to CD3/TCR-mediated or mitogenic stimulation. However, at cessation of treatment, when the antibody is cleared, the T cells rapidly restore the receptor on the cell surface [42].
In NOD mice presenting with recent onset diabetes, there is an almost complete clearing of insulitis within the first days of CD3 antibody treatment, coinciding with a rapid return to normoglycemia. T cells from treated mice exhibit a transient Th2 polarization, as assessed by increased production of IL-4. Such a Th2 shift does not appear to be causally linked to the therapeutic effect; in IL-4-deficient (IL-4-/-) NOD mice, CD3 antibody treatment induces disease remission and restoration of self-tolerance as efficiently as in wild-type mice [34].
Importantly, diabetogenic cells disappear during treatment, but recover in tolerant hosts, albeit in a variable fashion, depending on their fine specificity. Thus, our recent data show that, at least within the pathogenic CD8 compartment, preproinsulin-specific cells recover more rapidly and more efficiently than IGRP-specific cells [50]. During this recovery phase, by 2-6 weeks following the end of treatment, mononuclear cell infiltrates are observed in tolerant NOD mice. However, these cell infiltrates remain confined to the periphery of the islets (peripheral insulitis), and are not associated with active destruction of insulin-secreting β-cells. Concomitantly, a state of immunologically 'active' and transferable tolerance develops, involving TGFβ-dependent Tregs [34, 50]. The key role of TGFβ in the CD3 antibody-induced tolerance is well illustrated by its blockade, following the in vivo administration of a neutralizing TGFβ antibody [34, 50].
We have established a new preclinical model to assess the potency of potential therapeutic antibodies to human CD3. As mentioned above, the recognition of anti-human CD3 antibodies is restricted to human and chimpanzee T cells; they do not cross-react with lymphocytes from other species. Consequently, we have derived NOD mice expressing the human CD3ε chain as a transgene. The T cells of these mice are sensitive to anti-human CD3 antibodies in vitro and in vivo [50]. The data obtained from this model to date indicate that it appears to be a powerful tool to obtain further insight into the mode of action of anti-human CD3 antibodies, and to implement suitable treatment protocols, in particular those addressing combination therapies. Indeed, as compared to conventional NOD mice, human CD3ε NOD mice will allow the testing of the same antibodies that are used in the clinic.
3. Clinical trials using CD3 monoclonal antibodies in autoimmune diabetes
Based on the preclinical results in NOD mice, it was logical to evaluate the effectiveness of CD3 antibodies in patients presenting with recent onset autoimmune diabetes. Of course, the major concern was the potential toxicity, as witnessed with the first generation CD3 antibodies such as OKT3, linked to their mitogenic potential and related cytokine release. Therefore, attention was focused on humanized CD3 monoclonal antibodies that were mutated in their Fc portion to decrease binding to Fc receptors. These antibodies consequently minimize the cross-linking of T cell receptors that are known to drive T cell activation and cytokine release. Thus, the mitogenic response varied in vitro and in vivo, depending on the isotype of the murine CD3-specific antibody (IgG2a >> IgG1 >> IgG2b >> IgA), and on the CD3-specific F(ab')2 fragments that lack the Fc portion are non-mitogenic [41, 51-52].
We will now discuss the clinical results obtained by using two humanized complementarity-determining region (CDR)-grafted CD3 antibodies, which have been the subject of clinical trials conducted in autoimmune diabetes. These are:
1. ChAglyCD3 (otelixizumab), derived from the rat YTH 12.5 antibody, which expresses a single mutation that prevents glycosylation of its γ1 Fc portion [53].
2. OKT3γ1 Ala-Ala (teplizumab), the humanized version of OKT3, a human IgG1 with two mutations in its Fc portion [54].
Phase I safety trials were carried out with the two antibodies in renal allograft recipients presenting with acute rejection episodes. The results confirmed that their administration did not elicit major side effects [55-56].
In autoimmune diabetes, two academic trials were launched simultaneously in the year 2000. A phase I open trial using teplizumab for 14 days included a total of 48 patients (24 antibody-treated and 24 untreated) [57-58]. The results suggested that in antibody-treated patients the progression of disease had been satisfactorily restrained for more than one year, as assessed by C-peptide production following mixed meal test stimulation and exogenous insulin requirements.
In parallel, in a multinational European collaborative effort involving clinicians and biologists from Belgium, Germany, France, and the UK, we conducted a phase II randomized, double-blind, controlled trial including 80 patients who were randomized to receive otelixizumab (40 patients) or placebo (40 patients) [16-17]. A dose of 8 mg of the antibody or placebo was injected i.v. for 6 days (48 mg cumulated dose). Data were reported at 18 months and 48 months of follow-up; at that time, a total of 63 patients evenly distributed between the two groups were available for analysis. The data demonstrated that otelixizumab treatment preserved the insulin-secreting capacity of β-cells very efficiently. This was assessed at 6, 12, and 18 months by measuring C-peptide after controlled i.v. glucose stimulation during a clamp test [16]. Concomitantly, a significant decrease in insulin needs was observed in the otelixizumab-treated patients, as compared to placebo. Remarkably, at 18 months, 75% of the treated patients, harboring the highest β-cell mass at the time of inclusion (higher than the median value of the whole population), showed insulin needs ≤0.25 U/kg/day, a threshold normally regarded as the hallmark of clinical insulin independency. None of the placebo-treated patients had these low insulin needs. At 48 months, the insulin needs were still significantly different in otelixizumab- versus placebo-treated patients. However, the therapeutic effect began to disappear by 24 months, as judged by comparing the slopes of the curves [17].
Relatively minor acute side effects, linked to a persisting, though limited, cytokine release, were observed after the first infusion. However, these side effects did not necessitate any pretreatment nor did they hamper the normal enrolment of patients. Transient reactivation of Epstein Barr virus (EBV) was observed, as assessed by an increase in the number of EBV copies 10-20 days after the first injection, measured in peripheral blood mononuclear cells using quantitative PCR. By 3 weeks, in all patients, the number of EBV copies returned to normal baseline pretreatment levels, presumably following an efficient humoral and cellular immune response specific to EBV development [59]. In long-term follow-up, none of the patients exhibited a biological sign of viral reactivation or related clinical morbidity. The importance of this observation in terms of safety is obvious. These data should be considered in the context of the recent results, showing that EBV serologic reactivation, with no clinical morbidity, has also been observed in patients with early onset T1D treated with the CD25-specific monoclonal antibodies daclizumab and mycophenolate mofetil [60]. Furthermore, the data suggest that the effect of otelixizumab in patients is antigen-specific, i.e. the antibody treatment affects the autoimmune reaction, but does not impair immune responses to unrelated exogenous antigens, as observed in NOD mice.
For each of the two monoclonal antibodies, phase III trials were initiated and were termed "DEFEND-1" for otelixizumab, and "Protégé" for teplizumab. DEFEND-1 was a randomized, placebo-controlled study including 272 patients, aged 12 to 45, with new-onset T1D treated with insulin for not more than 3 months. The patients received a single 8-day i.v. course of a cumulated dose of 3.1 mg of otelixizumab (a dose well below that used in our academic trial). In March 2011, a press release announced that the primary endpoint (a difference in the levels of C-peptide following a mixed meal test at 12 months after treatment) had not been met (http://www.gsk.com/media-news/pressre- leases/2011/2011_pressrelease_10039.htm). Due to these findings the enrolment in the other phase III trial using otelixizumab, DEFEND-2, was stopped.
Protégé was designed as a randomized placebo-controlled study including 554 patients, aged 8-35, with new-onset T1D treated with insulin for not more than 3 months. The patients received a 14-day course twice at day 0, and 6 months with 3 different dose regimens (cumulated dose 5, 6, or 17 mg x 2). The composite end point included HbA1c <6.5% and insulin <0.5 U/kg/day. In October 2010, a press release announced that the primary end point, which in this case was the composite including a fixed HbA1C value (<6.5%) and exogenous insulin needs at 12 months (<0.5 U/kg/day), had not been met (http://www.macrogenics.com/ press_releases-284.html). Due to this result the enrolment in the other phase III trial using teplizumab, Protégé Encore was stopped.
However, a further post-hoc analysis, using conventional end-points validated by all the previous trials namely, C-peptide production and insulin needs, concluded that a significant therapeutic effect had been achieved [61]. Importantly, as seen in the results of the academic placebo-controlled trial with otelixizumab, the effect was more pronounced in patients with the highest stimulated C-peptide at inclusion. Moreover, it appeared that the effect was more pronounced in children. As expected, the effect was dose-dependent; only patients who received the cumulative dose of 17 mg showed a beneficial effect.
4. Conclusions: is the glass half empty or half full?
In contrast to promising academic studies, the phase III trials with teplizumab and otelixizumab generated a different outcome. It is important to clarify the reasons for this discrepancy. Therefore, we aim to summarize for our readers what we think are the key features that resulted in so-called 'failed' phase III trials.
It may well be that the pharmaceutical development proceeded too rapidly. Normally, one would have hoped to follow those first academic trials with proper phase II trials to select optimal dosing strategies, seeking the ideal balance between safety and therapeutic efficacy. In the case of otelixizumab, the chosen dose, aimed at minimizing side effects, resulted in a protocol with a dose too low for clinical efficacy. As a result, the phase III trial utilized a dose 16 times lower than the effective dose used in the previously successful academic trial! In the other case, the basic parameters necessary to define the adequate dose of teplizumab were available, but data analysis avoided the power of stimulated C-peptide measures, and ended with a non-validated composite end point.
With these explanations in mind, the reality is that patients have been successfully treated in the initial clinical trials, but phase III trials failed to prove efficacy. The data can be summarized as follows:
1. Several hundred patients received a therapeutic dose of a CD3 antibody.
2. Safety data appeared to be acceptable within the dose parameters used.
3. Clinical efficacy in one study was observed over a long period of time following a very short treatment, consistent with clinical operational tolerance.
At this time, no other drug tested in T1D had offered such results. One exception being the data reported in a press release by Andromeda on the use of the autoantigen diapep277 (http://www.rttnews.com/1766892/andromeda-s-type-1-diabetes-drug-diapep277-meets-primary-endpoint-in-phase-3.aspx).
By no means do we imagine that CD3 antibodies, especially if used alone, could be the ultimate solution [62]. Of course, as in all complex diseases, we have to seriously consider combination strategies, for which encouraging experimental data exists [63-64]. We also need to define patients who may be good responders to immune therapy, which requires the identification of suitable biomarkers. We should also consider treating patients earlier, in prevention studies, before too significant damage is inflicted upon β-cells, as is being attempted by TrialNet (J. Skyler, personal communication). But above all, we have a collective responsibility, beyond individual interests, to encourage the development of any strategy that shows the potential to induce durable remission of established T1D, as it is the case for CD3 antibodies.
Disclosure: LC has no conflict of interest to disclose. HW is the originator of otelixuzumab for which royalty sharing arrangements exist with Cambridge and Oxford Universities.
References
- Castano L, Eisenbarth GS. Type-I diabetes: a chronic autoimmune disease of human, mouse, and rat. Annu Rev Immunol 1990. 8:647-679. [DOD] [CrossRef]
- Bach JF. Insulin-dependent diabetes mellitus as an autoimmune disease. Endocrine Rev 1994. 15:516-542. [DOD]
- Eizirik DL, Colli ML, Ortis F. The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat Rev Endocrinol 2009. 5:219-226. [DOD] [CrossRef]
- Harjutsalo V, Sjoberg L, Tuomilehto J. Time trends in the incidence of type 1 diabetes in Finnish children: a cohort study. Lancet 2008. 371:1777-1782. [DOD] [CrossRef]
- Katz JD, Wang B, Haskins K, Benoist C, Mathis D. Following a diabetogenic T cell from genesis through pathogenesis. Cell 1993. 74:1089-1100. [DOD] [CrossRef]
- Billiard F, Litvinova E, Saadoun D, Djelti F, Klatzmann D, Cohen JL, Marodon G, Salomon BL. Regulatory and effector T cell activation levels are prime determinants of in vivo immune regulation. J Immunol 2006. 177:2167-2174. [DOD]
- Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, Bluestone JA. B7/CD28 Costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity 2000. 12:431-440. [DOD] [CrossRef]
- You S, Belghith M, Cobbold S, Alyanakian MA, Gouarin C, Barriot S, Garcia C, Waldmann H, Bach JF, Chatenoud L. Autoimmune diabetes onset results from qualitative rather than quantitative age-dependent changes in pathogenic T-cells. Diabetes 2005. 54:1415-1422. [DOD] [CrossRef]
- You S, Leforban B, Garcia C, Bach JF, Bluestone JA, Chatenoud L. Adaptive TGF-{beta}-dependent regulatory T cells control autoimmune diabetes and are a privileged target of anti-CD3 antibody treatment. Proc Natl Acad Sci U S A 2007. 104:6335-6340. [DOD] [CrossRef]
- Kurrer MO, Pakala SV, Hanson HL, Katz JD. Beta cell apoptosis in T cell-mediated autoimmune diabetes. Proc Natl Acad Sci USA 1997. 94:213-218. [DOD] [CrossRef]
- Lebastchi J, Deng S, Lebastchi AH, Beshar I, Gitelman S, Willi S, Gottlieb P, Akirav EM, Bluestone JA, Herold KC. Immune therapy and beta-cell death in type 1 diabetes. Diabetes 2013. In press. [DOD]
- Strandell E, Eizirik DL, Sandler S. Reversal of beta-cell suppression in vitro in pancreatic islets isolated from nonobese diabetic mice during the phase preceding insulin-dependent diabetes mellitus. J Clin Invest 1990. 85:1944-1950. [DOD] [CrossRef]
- Sreenan S, Pick AJ, Levisetti M, Baldwin AC, Pugh W, Polonsky KS. Increased beta-cell proliferation and reduced mass before diabetes onset in the nonobese diabetic mouse. Diabetes 1999. 48:989-996. [DOD] [CrossRef]
- Clevers H, Alarcon B, Wileman T, Terhorst C. The T cell receptor/CD3 complex: a dynamic protein ensemble. Annu Rev Immunol 1988. 6:629-662. [DOD] [CrossRef]
- Chatenoud L, Thervet E, Primo J, Bach JF. Anti-CD3 antibody induces long-term remission of overt autoimmunity in nonobese diabetic mice. Proc Natl Acad Sci USA 1994. 91:123-127. [DOD] [CrossRef]
- Keymeulen B, Vandemeulebroucke E, Ziegler AG, Mathieu C, Kaufman L, Hale G, Gorus F, Goldman M, Walter M, Candon S, et al. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med 2005. 352:2598-2608. [DOD] [CrossRef]
- Keymeulen B, Walter M, Mathieu C, Kaufman L, Gorus F, Hilbrands R, Vandemeulebroucke E, Van de Velde U, Crenier L, De Block C, Candon S, Waldmann H, Ziegler AG, Chatenoud L, Pipeleers D. Four-year metabolic outcome of a randomised controlled CD3-antibody trial in recent-onset type 1 diabetic patients depends on their age and baseline residual beta cell mass. Diabetologia 2010. 53:614-623. [DOD] [CrossRef]
- Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975. 256:495-497. [DOD] [CrossRef]
- Kung P, Goldstein G, Reinherz EL, Schlossman SF. Monoclonal antibodies defining distinctive human T cell surface antigens. Science 1979. 206:347-349. [DOD] [CrossRef]
- Cosimi AB, Colvin RB, Burton RC, Rubin RH, Goldstein G, Kung PC, Hansen WP, Delmonico FL, Russell PS. Use of monoclonal antibodies to T-cell subsets for immunologic monitoring and treatment in recipients of renal allografts. N Engl J Med 1981. 305:308-314. [DOD] [CrossRef]
- Ortho Multicenter Transplant Study Group. A randomized clinical trial of OKT3 monoclonal antibody for acute rejection of cadaveric renal transplants. N Engl J Med 1985. 313:337-342. [DOD] [CrossRef]
- Vigeral P, Chkoff N, Chatenoud L, Campos H, Lacombe M, Droz D, Goldstein G, Bach JF, Kreis H. Prophylactic use of OKT3 monoclonal antibody in cadaver kidney recipients. Utilization of OKT3 as the sole immunosuppressive agent. Transplantation 1986. 41:730-733. [DOD] [CrossRef]
- Salmeron A, Sanchez-Madrid F, Ursa MA, Fresno M, Alarcon B. A conformational epitope expressed upon association of CD3-epsilon with either CD3-delta or CD3-gamma is the main target for recognition by anti-CD3 monoclonal antibodies. J Immunol 1991. 147(9):3047-3052. [DOD]
- Rao PE, Olini G, Kille J, Muchmore E, Talle MA, Brake G, Rudnick SA. OKT3E, an anti-CD3 antibody that does not elicit side effects or antiidiotype responses in chimpanzees. Transplantation 1991. 52:691-697. [DOD] [CrossRef]
- Van Wauwe JP, De Mey JR, Goossens JG. OKT3: a monoclonal anti-human T lymphocyte antibody with potent mitogenic properties. J Immunol 1980. 124:2708-2713. [DOD]
- Chatenoud L, Ferran C, Legendre C, Thouard I, Merite S, Reuter A, Gevaert Y, Kreis H, Franchimont P, Bach JF. In vivo cell activation following OKT3 administration. Systemic cytokine release and modulation by corticosteroids. Transplantation 1990. 49:697-702. [DOD] [CrossRef]
- Abramowicz D, Schandene L, Goldman M, Crusiaux A, Vereerstraeten P, De Pauw L, Wybran J, Kinnaert P, Dupont E, Toussaint C. Release of tumor necrosis factor, interleukin-2, and gamma-interferon in serum after injection of OKT3 monoclonal antibody in kidney transplant recipients. Transplantation 1989. 47:606-608. [DOD] [CrossRef]
- Herbelin A, Chatenoud L, Roux-lombard P, De Groote D, Legendre C, Dayer JM, Descamps-latscha B, Kreis H, Bach JF. In vivo soluble tumor necrosis factor receptor release in OKT3-treated patients. Differential regulation of TNF-sR55 and TNF-sR75. Transplantation 1995. 59:1470-1475. [DOD] [CrossRef]
- Leo O, Foo M, Sachs DH, Samelson LE, Bluestone JA. Identification of a monoclonal antibody specific for a murine T3 polypeptide. Proc Natl Acad Sci USA 1987. 84:1374-1378. [DOD] [CrossRef]
- Nicolls MR, Aversa GG, Pearce NW, Spinelli A, Berger MF, Gurley KE, Hall BM. Induction of long-term specific tolerance to allografts in rats by therapy with an anti-CD3-like monoclonal antibody. Transplantation 1993. 55:459-468. [DOD] [CrossRef]
- Tomonari K. A rat antibody against a structure functionally related to the mouse T-cell receptor/T3 complex. Immunogenetics 1988. 28:455-458. [DOD] [CrossRef]
- Plain KM, Chen J, Merten S, He XY, Hall BM. Induction of specific tolerance to allografts in rats by therapy with non-mitogenic, non-depleting anti-CD3 monoclonal antibody: association with TH2 cytokines not anergy. Transplantation 1999. 67:605-613. [DOD] [CrossRef]
- Hayward AR, Shreiber M. Neonatal injection of CD3 antibody into nonobese diabetic mice reduces the incidence of insulitis and diabetes. J Immunol 1989. 143:1555-1559. [DOD]
- Belghith M, Bluestone JA, Barriot S, Megret J, Bach JF, Chatenoud L. TGF-beta-dependent mechanisms mediate restoration of self-tolerance induced by antibodies to CD3 in overt autoimmune diabetes. Nat Med 2003. 9:1202-1208. [DOD] [CrossRef]
- Chatenoud L. CD3-specific antibody-induced active tolerance: from bench to bedside. Nat Rev Immunol 2003. 3:123-132. [DOD] [CrossRef]
- Chatenoud L, Primo J, Bach JF. CD3 antibody-induced dominant self tolerance in overtly diabetic NOD mice. J Immunol 1997. 158:2947-2954. [DOD]
- Kohm AP, Williams JS, Bickford AL, McMahon JS, Chatenoud L, Bach JF, Bluestone JA, Miller SD. Treatment with nonmitogenic anti-CD3 monoclonal antibody induces CD4+ T cell unresponsiveness and functional reversal of established experimental autoimmune encephalomyelitis. J Immunol 2005. 174:4525-4534. [DOD]
- Perruche S, Zhang P, Liu Y, Saas P, Bluestone JA, Chen W. CD3-specific antibody-induced immune tolerance involves transforming growth factor-beta from phagocytes digesting apoptotic T cells. Nat Med 2008. 14:528-535. [DOD] [CrossRef]
- Ochi H, Abraham M, Ishikawa H, Frenkel D, Yang K, Basso AS, Wu H, Chen ML, Gandhi R, Miller A, Maron R, Weiner HL. Oral CD3-specific antibody suppresses autoimmune encephalomyelitis by inducing CD4(+)CD25(-)LAP(+) T cells. Nat Med 2006. 12:627-635. [DOD] [CrossRef]
- Ludviksson BR, Ehrhardt RO, Strober W. TGF-beta production regulates the development of the 2,4,6-trinitrophenol-conjugated keyhole limpet hemocyanin-induced colonic inflammation in IL-2-deficient mice. J Immunol 1997. 159:3622-3628. [DOD]
- Hirsch R, Bluestone JA, De Nenno L, Gress RE. Anti-CD3 F(ab')2 fragments are immunosuppressive in vivo without evoking either the strong humoral response or morbidity associated with whole mAb. Transplantation 1990. 49:1117-1123. [DOD] [CrossRef]
- Chatenoud L, Baudrihaye MF, Kreis H, Goldstein G, Schindler J, Bach JF. Human in vivo antigenic modulation induced by the anti-T cell OKT3 monoclonal antibody. Eur J Immunol 1982. 12:979-982. [DOD] [CrossRef]
- Buysmann S, Bemelman FJ, Schellekens PT, Van Kooyk Y, Figdor CG, Ten Berge IJ. Activation and increased expression of adhesion molecules on peripheral blood lymphocytes is a mechanism for the immediate lymphocytopenia after administration of OKT3. Blood 1996. 87:404-411. [DOD]
- Wong JT, Colvin RB. Selective reduction and proliferation of the CD4+ and CD8+ T cell subsets with bispecific monoclonal antibodies: evidence for inter-T cell-mediated cytolysis. Clin Immunol Immunopathol 1991. 58:236-250. [DOD] [CrossRef]
- Wesselborg S, Janssen O, Kabelitz D. Induction of activation-driven death (apoptosis) in activated but not resting peripheral blood T cells. J Immunol 1993. 150:4338-4345. [DOD]
- Penaranda C, Tang Q, Bluestone JA. Anti-CD3 therapy promotes tolerance by selectively depleting pathogenic cells while preserving regulatory T cells. J Immunol 2011. 187:2015-2022. [DOD] [CrossRef]
- You S, Zuber J, Kuhn C, Baas M, Valette F, Sauvaget V, Sarnacki S, Sawitzki B, Bach JF, Volk HD, Chatenoud L. Induction of allograft tolerance by monoclonal CD3 antibodies: a matter of timing. Am J Transplant 2012. 12:2909-2919. [DOD] [CrossRef]
- Chatenoud L. Immune therapy for type 1 diabetes mellitus-what is unique about anti-CD3 antibodies? Nat Rev Endocrinol 2010. 6:149-157. [DOD]
- Chatenoud L, Bach JF. Antigenic modulation: a major mechanism of antibody action. Immunol Today 1984. 5:20-25. [DOD] [CrossRef]
- Kuhn C, You S, Valette F, Hale G, van Endert P, Bach JF, Waldmann H, Chatenoud L. Human CD3 transgenic mice: preclinical testing of antibodies promoting immune tolerance. Sci Transl Med 2011. 3:68ra10. [DOD] [CrossRef]
- Van Lier RA, Boot JH, De Groot ER, Aarden LA. Induction of T cell proliferation with anti-CD3 switch-variant monoclonal antibodies: effects of heavy chain isotype in monocyte-dependent systems. Eur J Immunol 1987. 17:1599-1604. [DOD] [CrossRef]
- Parlevliet KJ, Ten Berge IJ, Yong SL, Surachno J, Wilmink JM, Schellekens PT. In vivo effects of IgA and IgG2a anti-CD3 isotype switch variants. J Clin Invest 1994. 93:2519-2525. [DOD] [CrossRef]
- Bolt S, Routledge E, Lloyd I, Chatenoud L, Pope H, Gorman SD, Clark M, Waldmann H. The generation of a humanized, non-mitogenic CD3 monoclonal antibody which retains in vitro immunosuppressive properties. Eur J Immunol 1993. 23:403-411. [DOD] [CrossRef]
- Alegre ML, Peterson LJ, Xu D, Sattar HA, Jeyarajah DR, Kowalkowski K, Thistlethwaite JR, Zivin RA, Jolliffe L, Bluestone JA. A non-activating "humanized" anti-CD3 monoclonal antibody retains immunosuppressive properties in vivo. Transplantation 1994. 57:1537-1543. [DOD]
- Friend PJ, Hale G, Chatenoud L, Rebello P, Bradley J, Thiru S, Phillips JM, Waldmann H. Phase I study of an engineered aglycosylated humanized CD3 antibody in renal transplant rejection. Transplantation 1999. 68:1632-1637. [DOD] [CrossRef]
- Woodle ES, Xu D, Zivin RA, Auger J, Charette J, O'Laughlin R, Peace D, Jollife LK, Haverty T, Bluestone JA, Thistlethwaite JR Jr. Phase I trial of a humanized, Fc receptor nonbinding OKT3 antibody, huOKT3gamma1(Ala-Ala) in the treatment of acute renal allograft rejection. Transplantation 1999. 68(5):608-616. [DOD] [CrossRef]
- Herold KC, Hagopian W, Auger JA, Poumian Ruiz E, Taylor L, Donaldson D, Gitelman SE, Harlan DM, Xu D, Zivin RA, Bluestone JA. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med 2002. 346:1692-1698. [DOD] [CrossRef]
- Herold KC, Gitelman SE, Masharani U, Hagopian W, Bisikirska B, Donaldson D, Rother K, Diamond B, Harlan DM, Bluestone JA. A single course of anti-CD3 monoclonal antibody hOKT3gamma1(Ala-Ala) results in improvement in C-peptide responses and clinical parameters for at least 2 years after onset of type 1 diabetes. Diabetes 2005. 54:1763-1769. [DOD] [CrossRef]
- Keymeulen B, Candon S, Fafi-Kremer S, Ziegler A, Leruez-Ville M, Mathieu C, Vandemeulebroucke E, Walter M, Crenier L, Thervet E, et al. Transient Epstein-Barr virus reactivation in CD3 monoclonal antibody-treated patients. Blood 2010. 115:1145-1155. [DOD] [CrossRef]
- Loechelt BJ, Boulware D, Green M, Baden LR, Gottlieb P, Krause-Steinrauf H, Weinberg A. Epstein-Barr and other herpesvirus infections in patients with early onset type 1 diabetes treated with daclizumab and mycophenolate mofetil. Clin Infect Dis 2013. 56:248-254. [DOD] [CrossRef]
- Sherry N, Hagopian W, Ludvigsson J, Jain SM, Wahlen J, Ferry RJ Jr, Bode B, Aronoff S, Holland C, Carlin D, et al. Teplizumab for treatment of type 1 diabetes (Protege study): 1-year results from a randomised, placebo-controlled trial. Lancet 2011. 378(9790):487-497. [DOD] [CrossRef]
- Staeva TP, Chatenoud L, Insel R, Atkinson MA. Recent lessons learned from prevention and recent-onset type 1 diabetes immunotherapy trials. Diabetes 2013. 62:9-17. [DOD] [CrossRef]
- Bresson D, Togher L, Rodrigo E, Chen Y, Bluestone JA, Herold KC, von Herrath M. Anti-CD3 and nasal proinsulin combination therapy enhances remission from recent-onset autoimmune diabetes by inducing Tregs. J Clin Invest 2006. 116:1371-1381. [DOD] [CrossRef]
- Ablamunits V, Henegariu O, Hansen JB, Opare-Addo L, Preston-Hurlburt P, Santamaria P, Mandrup-Poulsen T, Herold KC. Synergistic reversal of type 1 diabetes in NOD mice with anti-CD3 and interleukin-1 blockade: evidence of improved immune regulation. Diabetes 2012. 61:145-154. [DOD] [CrossRef]
This article has been cited by other articles:

|
Invasive fungal infections in paediatric patients treated with macromolecular immunomodulators other than tumour necrosis alpha inhibitors
Kyriakidis I, Tragiannidis A, Zündorf I, Groll AH
Mycoses 2017. In press
|
|
|
Entire CD3ε, δ, and γ humanized mouse to evaluate human CD3-mediated therapeutics
Ueda O, Wada NA, Kinoshita Y, Hino H, Kakefuda M, Ito T, Fujii E, Noguchi M, Sato K, Morita M, Tateishi H, Matsumoto K, Goto C, Kawase Y, Kato A, Hattori K, Nezu J, Ishiguro T, Jishage KI
Sci Rep 2017. 7:45839
|
|

|
Mucosal administration of CD3-specific monoclonal antibody inhibits diabetes in NOD mice and in a preclinical mouse model transgenic for the CD3 epsilon chain
Kuhn C, Rezende RM, da Cunha AP, Valette F, Quintana FJ, Chatenoud L, Weiner HL
J Autoimmun 2016. In press
|
|

|
The homing and inhibiting effects of hNSCs-BMP4 on human glioma stem cells
Liu S, Yin F, Zhao M, Zhou C, Ren J, Huang Q, Zhao Z, Mitra R, Fan W, Fan M
Oncotarget 2016. 7(14):17920-17931
|
|

|
Therapeutic anti-CD3 monoclonal antibodies: from bench to bedside
Kuhn C, Weiner HL
Immunotherapy 2016. 8(8):889-906
|
|
|
Inducing and Administering Tregs to Treat Human Disease
Perdigoto AL, Chatenoud L, Bluestone JA, Herold KC
Front Immunol 2016. 6:654
|
|

|
Oral immune therapy: targeting the systemic immune system via the gut immune system for the treatment of inflammatory bowel disease
Ilan Y
Clin Transl Immunology 2016. 5(1):e60
|
|

|
Lung transplantation: chronic allograft dysfunction and establishing immune tolerance
Gracon AS, Wilkes DS
Hum Immunol 2014. 75(8):887-894
|
|

|
Type 1 diabetes therapy beyond T cell targeting: monocytes, B cells, and innate lymphocytes
Wong FS, Wen L
Rev Diabet Stud 2012. 9(4):289-304
|
|
|


)

