Chapter III. Re-establishing Tolerance
| Rev Diabet Stud,
2012,
9(4):348-356 |
DOI 10.1900/RDS.2012.9.348 |
In Vivo Delivery of Nucleic Acid-Formulated Microparticles as a Potential Tolerogenic Vaccine for Type 1 Diabetes
Valentina Di Caro1,2, Nick Giannoukakis1,3, Massimo Trucco1
1Division of Immunogenetics, Department of Pediatrics, Children´s Hospital of Pittsburgh, University of Pittsburgh School of Medicine, 4401 Penn Avenue, Pittsburgh, PA 15224, USA
2Ri.Med Foundation, via Bandiera 11, 90133, Palermo, Italy
3Department of Pathology, Children´s Hospital of Pittsburgh, University of Pittsburgh School of Medicine, 4401 Penn Avenue, Pittsburgh, PA 15224, USA
Address correspondence to: Massimo Trucco, e-mail mnt@pitt.edu
Manuscript submitted October 19, 2012; resubmitted October 31, 2012; accepted November 2, 2012.
Keywords: type 1 diabetes, immune tolerance , vaccination, autoimmunity, beta-cell, dendritic cell, antisense oligonucleotide, microparticles, regeneration
Abstract
Originally conceived as a method to silence transcription/translation of nascent RNA, nucleic acids aimed at downregulating gene expression have been shown to act at multiple levels. Some of the intriguing features of these gene-silencing nucleic acids include activation of molecular signals in immune cells which confer tolerogenic properties. We have discovered a method to induce stable tolerogenic ability to dendritic cells ex vivo using a mixture of phosphorothioate-modified antisense DNA targeting the primary transcripts of CD40, CD80 and CD86. Autologous human dendritic cells generated in the presence of these oligonucleotides prevent and reverse type 1 diabetes (T1D) in the non-obese diabetic (NOD) strain mouse model of the human disease, and have been shown to be safe in established diabetic human patients. Even though this ex vivo approach is clinically feasible, we have gone beyond a cell therapy approach to develop a "population-targeting" microsphere formulation of the three antisense oligonucleotides. Effectively, such a product could constitute an "off-the-shelf" vaccine. In this paper, we describe the progress made in developing this approach, as well as providing some insight into potential molecular mechanisms of action.
Abbreviations: AS-MSP – antisense oligonucleotide-formulated microsphere; BDCA – blood dendritic cell antigen; BIIB017 – PEGylated form of interferon beta-1a; Breg – regulatory B cell; CCL – chemokine (C-C motif) ligand; CCR – C-C chemokine receptor; CHS – contact hypersensitivity; CN – control ; DC – dendritic cell; DEC-205 – dendritic and epithelial cells, 205 kDa (CD205); DNA – deoxyribonucleic acid; FDA – Food and Drug Administration; Foxp3 – forkhead box P3; GITR – glucocorticoid-induced TNF receptor family-related; GMP – good manufacturing practice; IDO - indoleamine 2,3-dioxygenase; Ig – immunoglobulin; IL – interleukin; ILT – Ig-like transcript; LPS – lipopolysaccharides; MDA5 – melanoma differentiation-associated protein 5; MHC – major histocompatibility complex; NCT – national clinical trial; NF-κB – nuclear factor kappa B; NKT – naturalkiller T; NOD – non-obese diabetic; PEG – polyethylene glycol; pMHC-NP – peptide/major histocompatibility complex (nucleoprotein); RIG-1 – retinoic acid-inducible gene 1; RNA – ribonucleic acid; RNAi – RNA interference; RNase – ribonuclease; T1D – type 1 diabetes; TCR – T cell receptor; TGF-β – transforming growth factor beta; Th – T helper; TLR – toll-like receptor; TNF-α – tumor necrosis factor alpha; UPMC – University of Pittsburgh Medical Center
1. Introduction
Type 1 diabetes (T1D) is an autoimmune disease caused by the loss of insulin-producing β-cells below a critical mass necessary to maintain proper glucose metabolism consequent to chronic impairment. A combination of multilocus genetic susceptibility and exposure to environmental triggers facilitates a T-cell-driven process of islet inflammation, impairment and eradication [1]. Studies conducted in non-obese diabetic (NOD) mice, a strain which spontaneously develops diabetes of an autoimmune nature, have highlighted the critical role of the adaptive immune response in the pathogenesis of the disease [2]. β-cell death is first observed at 2-3 weeks of age, possibly as part of a natural remodeling and/or metabolic re-programming of β-cells. Alternatively, or in addition, β-cell death could occur as a consequence of host response to viral infection [3].
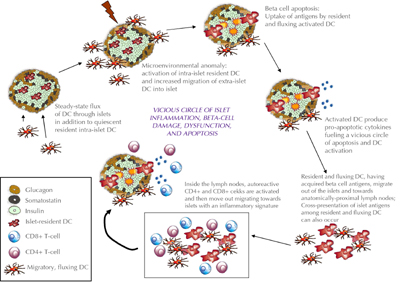
Irrespective of the actual initiating trigger, which is now considered to be insulin itself [4], the role of dendritic cells (DC) is critical. β-cell death leads to activation of islet-resident and pancreas-accumulating DCs in response to an as yet unidentified inflammatory signal deriving from the islets. Both islet-resident DCs and those that respond to an islet "damage" signal contribute to β-cell autoreactivity. They take up β-cell-derived material, which includes the antigens that drive the cell-specific inflammation before migrating to the pancreas-draining lymph-nodes, where they initiate the expansion of specific β-cell autoreactive T cells that have escaped negative thymic selection [5]. This cascade of events initiates a chronic inflammation and creates a vicious circle that eventually results in sufficient β-cell mass destruction to require exogenous insulin replacement, while peripheral mechanisms attempt to prevent autoimmunity, e.g. through Foxp3 T regulatory cells (Figure 1). Even if insulin replacement can maintain normal glucose homeostasis, it is unable to provide the physiologic control necessary to prevent serious diabetic complications such as cardiovascular diseases, nephropathy and neuropathy, which decrease quality of life and result in significant morbidity and mortality. Thus, insulin cannot cure T1D, but only mitigate the symptoms. An effective cure would need to preserve an adequate amount of the patients' residual β-cell mass to maintain physiologic glucoregulation, while eliminating or stably suppressing the underlying autoimmunity.
 |
 |
Figure 1. The autoimmune vicious circle favoring the spread of anti-β-cell epitope. Activated by an environmental stimulus, the autoreactive T cells that escaped thymic negative selection leave the lymph nodes and move into the tissues where they eventually find the self-peptide with which they were originally set up to react. Once the first β-cells are damaged, debris from dead cells are collected by DCs and presented to naïve T cells. T cells "ignorant" of the existence of these self-antigens, recognize them as foreign and react against them once back in the islet of Langerhans by killing new β-cells. This constitutes a vicious circle that does not allow the recovery of the insulin-secreting cells, even when the physiologic homeostasis process tries to substitute the lost cells with new cells. APC: antigen presenting cell. (From Phillips B, et al. A microsphere-based vaccine prevents and reverses new-onset autoimmune diabetes. Diabetes 2008. 57:1544.) [42]. |
|
There has been some success in achieving preservation of a residual β-cell mass which is able to regulate glucose homeostasis physiologically by using immunosuppressive drugs such as cyclosporin A and anti-CD3 antibody [6, 7]. However, the significant adverse events associated with these approaches require ongoing research activities to discover new and safer methods, including biologic or cell-related therapies aimed at mobilizing tolerogenic cell networks in vivo.
Immunotherapy to halt or reverse autoimmune diabetes needs to establish and maintain stable peripheral T cell tolerance. Given the important role of DCs in the initiation of immune responses, DC-based strategies seem to be attractive options to induce functional self-tolerance in T1D [8]. Studies involving DCs in therapeutic applications focus on the exogenous generation of tolerogenic DCs for administration as a cellular vaccine [9, 10]. An attractive alternative to ex vivo DC manipulation can be in vivo targeting of DCs with biologic or chemical drugs which stabilize the cells into tolerogenic states. Given the labile nature of biologics and most of the immunosuppressive chemicals, as well as their potential for systemic spread, biodegradable microparticles have evolved which have been shown to mitigate these unwanted effects. Additionally, microparticles can be formulated in a manner that can result in the co-delivery of immunosuppressive agents along with disease-relevant antigens [11]. Furthermore, microparticles can be engineered to be multifunctional and modular. For example, they can be coated with chemoattractants (e.g. CCL19, CCL20, CCL21) that specifically stimulate DC accumulation to the area of administration in vivo [12, 13]. Once taken up, the microparticles can release their tolerogenic payload with or without the provision of antigens. Finally, microparticles can also be programmed to release their various contents at different times after in vivo injection.
2. Dendritic cells as immunoregulators
The interest in the use of DCs as cellular therapy began with the identification of their role as the most potent antigen-presenting cells. This led to more than 50 clinical trials worldwide using DCs as adjuvant immunotherapy for many malignancies [10, 14-16]. The common characteristic of these DCs is their high expression of costimulation surface proteins like CD86, CD40, and OX40L, conferred during the generation process by the addition of immunostimulatory cytokines ex vivo or triggered after the generation process by the addition of nucleic acids. Intensive costimulation inside the lymph nodes draining the site of DC administration results in very robust Th1 type reactions and activation of naive and memory T cells. Following ex vivo generation, immunostimulatory DCs are poorly phagocytic and exhibit lower thresholds for TLR stimulation. They consequently express high levels of NF-κB, and much of this transcription factor is found in the nucleus compared to non-stimulatory DCs where NF-κB is mainly cytosolic. Immunostimulatory DCs produce significant levels of IL-12, IL-6, and TNF-α. Through these cytokines the DCs manage and maintain a proinflammatory cell loop at the lymph nodes that drain their site of administration. This loop includes the activation of naive and memory T cells, macrophages, NK cells, and B cells. Most immunostimulatory DC protocols involve the provision of antigens derived from the cells or tissues to which an immune response should be targeted. For example, in prostate cancer trials, DCs are often pulsed with tumor-derived antigens in the form of the specific patient's tumor lysate. This increases the probability that T cells reactive to tumor antigens in vivo will be preferentially expanded by the DCs as the tumor antigen is presented on class II MHC.
Eventually, it was appreciated that DCs play a substantial role as regulators of immunity by providing activation and maintenance signals for a variety of immunosuppressive cells [17, 18]. Largely acting on T cell populations (CD4, CD8), DC-related regulation of immunity also involves the stimulation of novel immune cells which are not well characterized (NKT cells, gamma-delta T cells, T-follicular helper cells, and T-follicular regulatory cells) [19, 20]. General key features of DCs that activate and maintain immunosuppressive states include:
- Low antigen-presentation capacity (low levels of class I and class II MHC expression on the cell surface)
- Low-to-absent co-stimulation ability
- Poor or absent allostimulatory ability to induce T cell proliferation in allogeneic mixed leukocyte culture or in antigen-specific recall responses
- Production of Th2 type cytokines and retinoic acid [21, 22]
DCs with immunosuppressive roles occur naturally in vivo and have been generated ex vivo to induce negative immunomodulation. The naturally occurring DCs have been characterized as CD11c+, CD11b+, CD8 alpha+, CD45RB+, BDCA4+ CD123+, CCR7+, and CCR9+. Other DCs of this kind have been characterized as CD83-, CD1a+, ILT2+, ILT3+, and ILT4+, and yet others exhibit CD200R3+ and CD49+. Some reports indicate that expression of indoleamine 2,3-dioxygenase (IDO) is characteristic of immunosuppressive DCs. However, a respectable body of data suggests that IDO expression is limited to very specific and limited populations which are possibly in a metastable developmental stage in vivo [23, 24]. Although DCs have a natural ability to switch between activating immunostimulatory and suppressive tolerogenic states in vivo, the microenvironment determines their phenotype and the stability of the phenotype. Understanding the microenvironmental signals that enforce this modulation is the key to acquiring insight into DC-related biological mechanisms and to discovering approaches to achieve stable tolerogenic characteristics in ex vivo generated DCs.
Tolerogenic DCs can be generated in vitro from bone marrow precursors of rodents, or blood monocytes from non-human primates and humans, by controlling their exposure to cytokines, growth factors, pharmacological agents, or by genetic engineering [25]. It is therefore reasonable to exploit these effects to formulate microparticles that could target DCs in vivo. Different studies have shown that a downregulated costimulation capacity in DCs can lead to a tolerogenic therapeutic outcome [26, 27]. Indeed, in states of impaired costimulation, functionally immature DCs can achieve long-term and stable allograft survival, and can also prevent autoimmune disease in animal models [28, 29]. Mechanistically, functionally immature DCs, with low to absent costimulatory molecule expression, have been shown to mediate peripheral tolerance by inducing T cell anergy. This was observed in the context of cell-cell contact and/or cytokine secretion, and promoted the function and the expansion of regulatory cell subsets such as Foxp3+CD25+CD4+ Treg and novel B regulatory cell populations [10, 30, 31].
There are different mutually non-exclusive mechanisms which explain how tolerogenic DCs (i.e. functionally immature cells) maintain an overall state of immune hyporesponsiveness, of which the induction of anergy is the most clearly understood. In the absence of costimulation, T cells that interact with antigen through class II MHC on costimulation-deficient DCs (e.g. low surface levels of CD86 and OX40L) cannot enter the cell cycle. Those T cells that enter the cell cycle, exhibit aborted G1-S phase transition.
Costimulation can be thought of as a means to increase the time of interaction between the T cell receptor complex and the antigen-MHC on the DC. The lower the costimulatory ability the shorter is the interaction between DC and T cell, and the less time the TCR has to interact with the MHC-peptide. Increasing evidence demonstrates that a critical threshold exists for the time that a TCR is engaged with the MHC to stabilize the TCR signaling complex and to initiate phosphorylation of TCR epsilon-associated proteins. The latter amplifies the initial phosphorylation cascade. This culminates in the activation of cyclin production, cyclin-dependent kinase activation, and transition into G1-S. When the time of TCR-MHC engagement does not meet the critical threshold (e.g. consequent to impaired costimulation), cell cycle entry is aborted, which results in anergy in the T cell. Anergic T cells obtain an antigen-specific immunosuppressive ability. Through mechanisms which are still unclear, they silence or impair bystander immune cells to enter into a proinflammatory state.
Functionally immature and tolerogenic DCs also increase the frequency of regulatory T and B cells. Although some studies suggest that DC-derived anti-inflammatory cytokines (e.g. IL-10 and TGF-β) and/or DC surface proteins (e.g. GITR) expand pre-existing Foxp3+ Tregs, the biological processes are still unclear. Interestingly, oligonucleotides, including RNAi and antisense DNA, have an intrinsic capacity to trigger tolerogenicity in DCs, in a manner that is independent of their expected molecular effects (e.g. RNase H-dependent cleavage of the primary transcript). It is highly probable that they act through alternative activation of oligonucleotide-sensing receptors such as TLR2, 3, 7, and 9, and RIG-1 and MDA-5 [32]. While TLR signaling in DCs frequently results in an immunostimulatory state, in some instances, TLR signaling can confer immunosuppressive abilities. We speculate that this is not a TLR-dependent phenomenon per se, but depends on the higher order structure of the oligonucleotides which determine how TLRs and which TLRs will transmit a "higher order"-dependent signal. Such a possibility would involve differential TLR signaling dependent on the topology of its ligand.
3. Tolerogenic dendritic cells as cell therapy for type 1 diabetes
Promising preclinical data were obtained from studies in NOD mice, showing that DCs generated in vitro in the presence of a mixture of CD40-, CD80-, and CD86-targeting antisense DNA can prevent and reverse the disease. This success led to a phase I clinical trial using autologous ex vivo engineered DCs from established diabetic patients (clinicaltrials.gov identifier NCT00445913), conducted at the University of Pittsburgh Medical Center (UPMC) [10]. The trial included adult volunteers, aged from 18 to 60 years, with documented evidence of insulin-requiring T1D of at least 5 years' duration. Leukocytes were obtained from the patients by apheresis. DCs were generated in the presence of antisense DNA, targeting the primary transcripts of CD40, CD80, and CD86 costimulatory genes under good manufacturing practices (GMP). These DCs expressed low levels of CD80, CD86, and CD40, and were then injected into the patients by intradermal/subcutaneous administration at an anatomical site proximal to the pancreas. The choice of this anatomic region for DC delivery was based on the location of the lymphatic conduits (microvessels) which drain the injection site and favor migration of the DCs into the pancreatic lymph nodes. Once inside the pancreatic lymph nodes, the tolerogenic DCs could interact from the injection site with either soluble β-cell-derived antigens from the inflamed islets. The DCs can also acquire β-cell antigens through cross-presenting islet-resident DCs that moved into the pancreatic lymph nodes subsequent to β-cell destruction [33]. Having acquired the β-cell antigens, the tolerogenic DCs can then suppress autoreactive T cells and stimulate suppressive immune cells inside the pancreatic lymph nodes, which may stop the vicious circle maintaining the islet-specific inflammation in the pancreas.
Our study, the first ever to use tolerogenic DCs in humans, provided some interesting observations in addition to demonstrating its complete safety. First, treatment of diabetic patients with non-manipulated DCs or DCs generated in the presence of antisense DNA resulted in a C-peptide level that became detectable in some patients. Whereas, previous to and at enrolment of the patients, the C-peptide level was not detectable. Moreover, DC administration was associated with an increased frequency of B220+CD11c- B cells that we discovered to be comprised of a potentially regulatory B cell population (Breg) [10].
Further characterization of this B cell population confirmed their immunosuppressive activity in vitro [10]. In contrast to other reported Breg cells [34, 35], this immunosuppressive ability is independent of IL-10 secretion [10]. Although Bregs have been characterized by various phenotypes (reviewed in [36]), much attention has recently focused on a rare splenic B lymphocyte population, CD19highCD1dhighCD5+, which is able to suppress experimental contact hypersensitivity (CHS) in an antigen-restricted and IL-10-dependent manner [35, 37, 38]. These cells represent about 1% of the total splenic B cells. Adoptive transfer of these B lymphocytes effectively reduces inflammation in recipient mice sensitized with the same, but not with a different chemicals (in a CHS model in vivo), indicating that the suppressive function was antigen-specific. These cells require IL-10 for their suppressive effect [35, 37, 38]. Immature B lymphocytes, presenting what is very likely a transitional B220highCD21+CD23+ phenotype, have been shown to suppress the adoptive transfer of diabetes into immunodeficient NOD mice with diabetogenic immune cells [39]. B lymphocytes stimulated with LPS are suppressive in NOD and prevent diabetes. This effect is largely mediated by TGF-β [40]. B-lymphocyte-receptor-stimulated B cells from NOD mice delay T1D in syngeneic prediabetic recipients in an IL-10-dependent manner [41]. Our recent data suggest that the production of retinoic acid by DCs could be the possible link that mechanistically connects tolerogenic DCs and Bregs. Retinoic acid increases the survival of Bregs in vitro, but not that of non-suppressive CD19+B220+ B cells. Therefore, Bregs involve a selective survival advantage induced by tolerogenic DCs, which could maintain and/or amplify a suppressive milieu that would also include Foxp3+ Tregs (Di Caro et al., manuscript submitted).
After demonstrating that tolerogenic DCs are safe and well tolerated in patients with established T1D, an imminent phase II clinical trial in new onset T1D patients is scheduled. In addition to increasing the frequency and function of immunoregulatory cell populations in new-onset T1D patients, this trial is intended to test the efficacy of the tolerogenic DCs in improving functional residual β-cell mass. Stopping the inflammation process that promotes the T-cell-mediated β-cell destruction at such a stage could be sufficient to promote the rescue or regeneration of the remaining insulin-producing β-cell mass in the pancreas.
4. Dendritic cell-targeting microparticles
Despite the encouraging outcome of the phase I clinical trial, and the optimistic expectation for phase II, there are also limiting aspects for possible application in new onset diabetic children. Firstly, the leukapheresis process is invasive. It usually takes 2 or 3 hours to obtain precursor cells for the generation of a sufficiently large number of DCs. Moreover, tolerogenic DCs need to be generated in a well-equipped GMP facility for the collection, generation, storage, and distribution of a clinical-grade cell product. To overcome these issues we have been concurrently pursuing an alternative method for stable generation of immature DCs directly in vivo using microspheres as delivery vehicles of the antisense DNA [36].
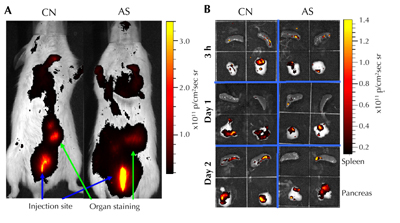
In the NOD mouse model, we have shown that these microspheres are phagocytosed by DCs, and confer a diabetes-suppressive phenotype on them. When injected, these microspheres mobilized endogenous DCs to the injection site, and within 3 hours the loaded DCs moved to the closest lymph nodes. Microsphere-administered mice remained diabetes-free when injected prior to disease, and at least 40% exhibited reversal of new onset disease. Diabetes-free mice exhibited an augmented Foxp3+CD25+CD4+ Treg cell frequency and hyporesponsiveness to β-cell antigens, without constraints on systematical immune responses to alloantigens. Additionally, T cells from successfully treated mice suppressed adoptive transfer of the disease by diabetogenic splenocytes into secondary immunodeficient NOD-SCID recipients. Finally, a fraction of the microspheres was found to be accumulated within the pancreas and the spleen, indicating uptake and migration by phagocytes. In vivo imaging measured the microsphere accumulation pattern (Figure 2). Based on these observations, we suggest that our "first-generation" microsphere formulation can be considered to be the first diabetes-suppressive vaccine to confer an immunoregulatory phenotype on endogenous DCs [42].
 |
|
Figure 2. In vivo accumulation of antisense-oligonucleotide-formulated microspheres (AS-MSP). A: Alive NOD mice received a subcutaneous injection containing sterile phosphate-buffered saline (control, CN) or fluorescent microspheres with 50 mg of AS-MSP (AS). Three hours post injection, the spheres accumulated in the area of the pancreas and spleen. B: Pancreas and spleen removed at 3, 24, and 48 h post injection contain the fluorescently labeled microspheres. (From Giannoukakis N, et al. Towards a cure for type 1 diabetes mellitus: diabetes-suppressive dendritic cells and beyond. Pediatr Diabetes 2008. 9(Part II):4 [44].) |
|
Other types of microparticle-based approaches have been explored as possible vaccines for T1D. Administration of T-cell-targeting nanoparticles coated with disease-relevant peptide/major histocompatibility complexes (pMHC-NPs) has been demonstrated to prevent T1D in prediabetic mice, and to restore normoglycemia in diabetic animals. Treatments of NOD mice, with specific pMHC-NPs, expand memory-like autoregulatory CD8+ T cells, and suppress local presentation of autoantigens [43]. Compared to ours, this approach is based on a different concept. While our approach envisages direct targeting of DCs to make them tolerogenic, pMHC-NPs trigger a pool of specific memory-like CD8+ T cells to modulate the autoimmune response. This includes suppression of the antigen-presenting function of DCs in an IFN-γ- and IDO-associated manner, and direct elimination of the antigen-loaded DCs via perforin [43]. Even though there is no obvious overlap in the biological mechanisms between the approaches, it is worth noting that the congruence of actions relates to the DCs.
Concerning the physical characteristics of micro-/nanoparticles, the most critical aspect is that the polymer carrier and chemical backbone is not immunogenic on its own, i.e. it does not stimulate immune cells through specific/non-specific processes. Most micro-/nanosphere formulations are comprised of three portions:
1. A hydrophilic polymer
2. A linker molecule
3. The biologically active compound
The properties of the polymer can be modified to affect the rate of drug clearance, solubility, immune responsiveness, and in vivo stability, facilitating decreased drug dosage and frequency of administration. Polyethylene glycol (PEG) is a non-toxic polymer which is a constituent of many clinical and nutritional products. Currently, two PEG conjugates have undergone clinical trials for the treatment of autoimmune diseases. In 1996, the interferon beta-1a drug Avonex was approved by the FDA for the treatment of multiple sclerosis. Phase III clinical trials with a PEGylated form of the drug designated BIIB017 are currently in progress. Another bioconjugate of PEG is an anti-tumor necrosis factor alpha (anti-TNF-α) antibody, initially tested for the treatment of rheumatoid arthritis and later extended to the treatment of Crohn's disease. The conjugate, termed Certolizumab pegol, was FDA-approved for the treatment of Crohn's disease in 2008 and rheumatoid arthritis in 2009. Despite the widespread effectiveness of PEG drug compounds, recent studies have found that their use can lead to the development of anti-PEG antibodies which may inhibit drug effectiveness. It needs to be clarified in future investigations whether this is a common problem not yet realized or specific only to certain PEG-compound formulations.
Apart from non-specific effects of formulation chemistry, the size of the particle decides whether DCs take it up by phagocytosis. Microspheres less than 200 microns in diameter are readily phagocytosed by macrophages and DCs. Interestingly, nanoparticles less than 50 microns in diameter are ignored by these cells. Thus, if the intention of the therapist is to take advantage of phagocytosis to deliver molecules inside DCs, then the critical diameter must be taken into account. If the intention is to modulate cell surface processes, the choice of the formulation size must exceed or be below the critical diameter (for example, using nanoparticles to target activation of surface receptors). Specific targeting of naturally occurring tolerogenic DCs in vivo may be required to avoid systemic immunosuppression. Even though there is no cell surface protein that selectively defines a tolerogenic DC, much evidence shows that DEC-205 is expressed on immature DCs in vivo. Conjugation of a DEC-205-targeting antibody to a microsphere formulation could be one means of ensuring preferential targeting of immature DCs in vivo.
5. Conclusion and perspective
Since T1D is a disease which largely affects children and adolescents, any therapy aimed at modifying the immune system towards re-establishment of physiologic glucoregulation would have to be minimally invasive, free of significant adverse events, and stably effective for at least a few months prior to a need to re-administer the agent(s). Considering the available clinical trial data, it seems that our approach of tolerogenic DCs is the safest with a promising potential to reverse autoimmune diabetes. We anticipate that adaptation of microparticle delivery will make the approach more appealing to patients, and will achieve the same biologic outcome with minimal logistical outlays and time from clinical confirmation of disease to initial therapy. Today, microspheres exist in numerous formulations that are inactive to the immune system (i.e., the particle chemistries do not induce non-specific immune activation). These very recent developments in polymer chemistry offer a variety of useful vehicles to target DCs in vivo.
As we approach the onset of a phase II clinical trial using the DCs, we are equally enthusiastic about the possibilities of a microparticle formulation for antisense DNA targeting CD40, CD80, and CD86, and for RNAi targeting of other costimulation pathways. The technology for verification of minimal off-target effects exists, and a careful selection of the most effective and specific oligonucleotide(s) coupled with disease-relevant antigen(s) could provide the elusive and long-sought T1D vaccine. By understanding how DCs process synthetic polymers, and how they respond to oligonucleotides, we are confident that this approach represents one of the safest ways of creating an off-the-shelf product. It is simple to manufacture to clinical grade on a large scale. The easy delivery to patients would make it possible to repeat the treatment by multiple administrations to achieve the final goal of reaching a complete tolerance against β-cell-specific antigens in the pancreas. It is also possible that these microparticles could represent a means for early intervention in first-degree relatives of T1D patients who exhibit signs of underlying disease (e.g. impaired first-phase insulin response to a mixed meal challenge along with high titers of T1D-related autoantibodies).
Disclosure: The authors report no conflict of interests.
Acknowledgments:
This work was supported in part by the Henry Hillman Endowment Chair of Pediatric Immunology awarded to Massimo Trucco.
References
- von Herrath M, Filippi C, Coppieters K. How viral infections enhance or prevent type 1 diabetes-from mouse to man. J Med Virol 2011. 83:1672. [DOD] [CrossRef]
- Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol 2005. 23:447-485. [DOD] [CrossRef]
- Conrad B, Weidmann E, Trucco G, Rudert WA, Behboo R, Ricordi C, Rodriquez-Rilo H, Finegold D, Trucco M. Evidence for superantigen involvement in insulin-dependent diabetes mellitus aetiology. Nature 1994. 371:351-355. [DOD] [CrossRef]
- Chentoufi AA, Polychronakos C. Insulin expression levels in the thymus modulate insulin-specific autoreactive T-cell tolerance: the mechanism by which the IDDM2 locus may predispose to diabetes. Diabetes 2002. 51:1383-1390. [DOD] [CrossRef]
- Fan Y, Rudert WA, Grupillo M, He J, Sisino G, Trucco M. Thymus-specific deletion of insulin induces autoimmune diabetes. Embo J 2009. 28:2812-2824. [DOD] [CrossRef]
- Di Caro V, Giannoukakis N, Trucco M. Tolerance and autoimmunity in type 1 diabetes. In: Mavragani CP (ed.). Autoimmune disorders - pathogenetic aspects. InTech, 2011, p. 193-220. [DOD]
- Chatenoud L, Bluestone JA. CD3-specific antibodies: a portal to the treatment of autoimmunity. Nat Rev Immunol 2007. 7:622-632. [DOD] [CrossRef]
- Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature 2007. 449:419-426. [DOD] [CrossRef]
- Hilkens CM, Isaacs JD, Thomson AW. Development of dendritic cell-based immunotherapy for autoimmunity. Int Rev Immunol 2010. 29:156-183. [DOD] [CrossRef]
- Giannoukakis N, Phillips B, Finegold D, Harnaha J, Trucco M. Phase I (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes Care 2011. 34:2026-2032. [DOD] [CrossRef]
- Keselowsky BG, Xia CQ, Clare-Salzler M. Multifunctional dendritic cell-targeting polymeric microparticles: engineering new vaccines for type 1 diabetes. Hum Vaccin 2011. 7:37-44. [DOD] [CrossRef]
- Zhao X, Jain S, Benjamin Larman H, Gonzalez S, Irvine DJ. Directed cell migration via chemoattractants released from degradable microspheres. Biomaterials 2005. 26:5048-5063. [DOD] [CrossRef]
- Singh A, Suri S, Roy K. In-situ crosslinking hydrogels for combinatorial delivery of chemokines and siRNA-DNA carrying microparticles to dendritic cells. Biomaterials 2009. 30:5187-5200. [DOD] [CrossRef]
- Pecher G, Haring A, Kaiser L, Thiel E. Mucin gene (MUC1) transfected dendritic cells as vaccine: results of a phase I/II clinical trial. Cancer Immunol Immunother 2002. 51:669-673. [DOD] [CrossRef]
- Su Z, Dannull J, Heiser A, Yancey D, Pruitt S, Madden J, Coleman D, Niedzwiecki D, Gilboa E, Vieweg J. Immunological and clinical responses in metastatic renal cancer patients vaccinated with tumor RNA-transfected dendritic cells. Cancer Res 2003. 63:2127-2133. [DOD]
- Palucka K, Ueno H, Fay J, Banchereau J. Harnessing dendritic cells to generate cancer vaccines. Ann N Y Acad Sci 2009. 1174:88-98. [DOD] [CrossRef]
- Steinman RM, Nussenzweig MC. Dendritic cells: features and functions. Immunol Rev 1980. 53:127-147. [DOD] [CrossRef]
- Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, Pulendran B, Palucka K. Immunobiology of dendritic cells. Annu Rev Immunol 2000. 18:767-811. [DOD] [CrossRef]
- Hegde S, Fox L, Wang X, Gumperz JE. Autoreactive natural killer T cells: promoting immune protection and immune tolerance through varied interactions with myeloid antigen-presenting cells. Immunology 2010. 130:471-483. [DOD] [CrossRef]
- Deenick EK, Ma CS, Brink R, Tangye SG. Regulation of T follicular helper cell formation and function by antigen presenting cells. Curr Opin Immunol 2011. 23:111-118. [DOD] [CrossRef]
- Coombes JL, Siddiqui KR, Arancibia-Carcamo CV, Hall J, Sun CM, Belkaid Y, Powrie F. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med 2007. 204:1757-1764. [DOD] [CrossRef]
- Sun CM, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR, Belkaid Y. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med 2007. 204:1775-1785. [DOD] [CrossRef]
- Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol 2004. 4:762-774. [DOD] [CrossRef]
- Baeke F, Takiishi T, Korf H, Gysemans C, Mathieu C. Vitamin D: modulator of the immune system. Curr Opin Pharmacol 2010. 10:482-496. [DOD] [CrossRef]
- Morelli AE, Thomson AW. Tolerogenic dendritic cells and the quest for transplant tolerance. Nat Rev Immunol 2007. 7:610-621. [DOD] [CrossRef]
- Nouri-Shirazi M, Thomson AW. Dendritic cells as promoters of transplant tolerance. Expert Opin Biol Ther 2006. 6:325-339. [DOD] [CrossRef]
- Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol 2003. 21:685-711. [DOD] [CrossRef]
- Chen D, Sung R, Bromberg JS. Gene therapy in transplantation. Transpl Immunol 2002. 9:301-314. [DOD] [CrossRef]
- Machen J, Harnaha J, Lakomy R, Styche A, Trucco M, Giannoukakis N. Antisense oligonucleotides down-regulating costimulation confer diabetes-preventive properties to nonobese diabetic mouse dendritic cells. J Immunol 2004. 173:4331-4341. [DOD]
- Giannoukakis N, Trucco M. A role for tolerogenic dendritic cell-induced B-regulatory cells in type 1 diabetes mellitus. Curr Opin Endocrinol Diabetes Obes 2012. 19:279-287. [DOD]
- Ueno H, Schmitt N, Palucka AK, Banchereau J. Dendritic cells and humoral immunity in humans. Immunol Cell Biol 2007. 88:376-380. [DOD] [CrossRef]
- Di Caro V, D'Anneo A, Phillips B, Engman C, Harnaha J, Trucco M, Giannoukakis N. Phosphatidylinositol-3-kinase activity during in vitro dendritic cell generation determines suppressive or stimulatory capacity. Immunol Res 2011. 50:130-152. [DOD] [CrossRef]
- Parish IA, Waithman J, Davey GM, Belz GT, Mintern JD, Kurts C, Sutherland RM, Carbone FR, Heath WR. Tissue destruction caused by cytotoxic T lymphocytes induces deletional tolerance. Proc Natl Acad Sci U S A 2009. 106:3901-3906. [DOD] [CrossRef]
- Iwata Y, Matsushita T, Horikawa M, Dilillo DJ, Yanaba K, Venturi GM, Szabolcs PM, Bernstein SH, Magro CM, Williams AD, et al. Characterization of a rare IL-10-competent B-cell subset in humans that parallels mouse regulatory B10 cells. Blood 2010. 117:530-541. [DOD] [CrossRef]
- DiLillo DJ, Matsushita T, Tedder TF. B10 cells and regulatory B cells balance immune responses during inflammation, autoimmunity, and cancer. Ann N Y Acad Sci 2010. 1183:38-57. [DOD] [CrossRef]
- Mauri C, Blair PA. Regulatory B cells in autoimmunity: developments and controversies. Nat Rev Rheumatol 2010. 6:636-643. [DOD] [CrossRef]
- Yanaba K, Bouaziz JD, Haas KM, Poe JC, Fujimoto M, Tedder TF. A regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T cell-dependent inflammatory responses. Immunity 2008. 28:639-650. [DOD] [CrossRef]
- Yanaba K, Bouaziz JD, Matsushita T, Tsubata T, Tedder TF. The development and function of regulatory B cells expressing IL-10 (B10 cells) requires antigen receptor diversity and TLR signals. J Immunol 2009. 182:7459-7472. [DOD] [CrossRef]
- Hu CY, Rodriguez-Pinto D, Du W, Ahuja A, Henegariu O, Wong FS, Shlomchik MJ, Wen L. Treatment with CD20-specific antibody prevents and reverses autoimmune diabetes in mice. J Clin Invest 2007. 117:3857-3867. [DOD] [CrossRef]
- Tian J, Zekzer D, Hanssen L, Lu Y, Olcott A, Kaufman DL. Lipopolysaccharide-activated B cells down-regulate Th1 immunity and prevent autoimmune diabetes in nonobese diabetic mice. J Immunol 2001. 167:1081-1089. [DOD]
- Hussain S, Delovitch TL. Intravenous transfusion of BCR-activated B cells protects NOD mice from type 1 diabetes in an IL-10-dependent manner. J Immunol 2007. 179:7225-7232. [DOD]
- Phillips B, Nylander K, Harnaha J, Machen J, Lakomy R, Styche A, Gillis K, Brown L, Lafreniere D, Gallo M, et al. A microsphere-based vaccine prevents and reverses new-onset autoimmune diabetes. Diabetes 2008. 57:1544-1555. [DOD] [CrossRef]
- Tsai S, Shameli A, Yamanouchi J, Clemente-Casares X, Wang J, Serra P, Yang Y, Medarova Z, Moore A, Santamaria P. Reversal of autoimmunity by boosting memory-like autoregulatory T cells. Immunity 2010. 32:568-580. [DOD] [CrossRef]
- Giannoukakis N, Phillips B, Trucco M. Toward a cure for type 1 diabetes mellitus: diabetes-suppressive dendritic cells and beyond. Pediatr Diabetes 2008. 9:4-13. [DOD] [CrossRef]
This article has been cited by other articles:
|


)
)


