Chapter III. Re-establishing Tolerance
| Rev Diabet Stud,
2012,
9(4):319-327 |
DOI 10.1900/RDS.2012.9.319 |
Tolerance Strategies Employing Antigen-Coupled Apoptotic Cells and Carboxylated PLG Nanoparticles for the Treatment of Type 1 Diabetes
Suchitra Prasad, Dan Xu, Stephen D. Miller
Department of Microbiology-Immunology and Interdepartmental Immunobiology Center, Feinberg School of Medicine, Northwestern University, Chicago, IL, USA
Address correspondence to: Stephen D. Miller, Department of Microbiology-Immunology, Feinberg School of Medicine, Northwestern University, 303 E. Chicago Avenue, Chicago, IL 60611, USA,, e-mail: s-d-miller@northwestern.edu
Manuscript submitted December 15, 2012; resubmitted January 8, 2013; accepted February 8, 2013.
Keywords: type 1 diabetes, immune tolerance, apoptosis, scavenger receptors, poly(lactide-co-glycolide), PLG nanoparticle, anergy, regulatory T cell, transplantation
Abstract
The development of therapies that specifically target autoreactive immune cells for the prevention and treatment of type 1 diabetes (T1D) without inducing generalized immunosuppression that often compromises the host's ability to clear non-self antigen is highly desired. This review discusses the mechanisms and potential therapeutic applications of antigen-specific T cell tolerance techniques using syngeneic apoptotic cellular carriers and synthetic nanoparticles that are covalently cross-linked to diabetogenic peptides or proteins through ethylene carbodiimide (ECDI) to prevent and treat T1D. Experimental models have demonstrated that intravenous injection of autoantigen decorated splenocytes and biodegradable nanoparticles through ECDI fixation effectively induce and maintain antigen-specific T cell abortive activation and anergy by T cell intrinsic and extrinsic mechanisms. The putative mechanisms include, but are not limited to, the uptake and processing of antigen-coupled nanoparticles or apoptotic cellular carriers for tolerogenic presentation by host splenic antigen-presenting cells, the induction of regulatory T cells, and the secretion of immune-suppressive cytokines, such as IL-10 and TGF-β. The safety profile and efficacy of this approach in preclinical animal models of T1D, including non-obese diabetic (NOD), BDC2.5 transgenic, and humanized mice, have been extensively investigated, and will be the focus of this review. Translation of this approach to clinical trials of T1D and other T cell-mediated autoimmune diseases will also be reviewed in this chapter.
Abbreviations: Ag – antigen; Ag-ECDI-SP – autoantigenic peptides chemically cross-linked to splenic leukocytes; APC - antigen-presenting cell; C57BL/6 – inbred strain C57 black 6; ChgA - chromogranin A; CTLA4 – cytotoxic T lymphocyte antigen 4; DC – dendritic cell; DTH – delayed-type hypersensitivity; EAE – experimental autoimmune encephalomyelitis; ECDI – ethylene carbodiimide; G6Pase – glucose 6-phosphatase; GAD65 – glutamate decarboxylase 65; GMP – good manufacturing practice; HLA – human leukocyte antigen; HSP60 – heat shock protein 60; IA-2 - islet cell antigen 512; IFN-γ – interferon gamma; IGRP – islet-specific G6Pase catalytic subunit-related protein; IL – interleukin; INS – insulin; ITN – Immune Tolerance Network; iTreg – induced regulatory T cell; i.v. – intravenous; MARCO - macrophage receptor with collagenous structure; MHC – major histocompatibility complex; MS – multiple sclerosis; MZM – marginal zone macrophage; NIT-1 – NOD-derived insulinoma cell line; NOD – non-obese diabetic; NOD.β2mnullHHD - HLA-A*0201 transgenic, H-2D(b)/mouse beta2-microglobulin double knockout mice; NRP - nonribosomal peptide; p31 - 1040-p31 peptide; PBL – peripheral blood leucocyte; PBMC – peripheral blood mononuclear cell; PD-1 – programmed cell death 1; PD-L1 – ligand of PD-1; PLG – poly(lactide-co-glycolide); PLGA – poly(lactic-co-glycolic) acid; PSB – polystyrene beads; RBC – red blood cell; T1D – type 1 diabetes; TCR – T cell receptor; Tg – transgenic; TGF-β – transforming growth factor beta; Th – T helper; Tr1 – type 1 regulatory T cell; Treg – regulatory T cell; UVB – ultraviolet B light
1. Introduction
The ability to tolerize T cells specific for autoantigens, allergens, and alloantigens remains the most desired treatment for a myriad of immune-mediated diseases, including autoimmune diseases such as type 1 diabetes (T1D). However, successful translation of tolerance-based immune therapies for treatment of autoimmune diseases to the clinic has yet to be realized. Thus far, therapies employing soluble Ag have had limited success in modulating T1D (reviewed in [1]). Subcutaneous injection of the 65-kDa isoform of glutamic acid decarboxylase in alum had no effect on disease progression [2]. Although mucosal antigen delivery has shown promise in animal models of multiple sclerosis and T1D [3], larger clinical trials testing oral and nasal administration of insulin have been ineffective in the prevention or reversal of new-onset T1D (reviewed in [4, 5]). Intravenous administration of humanized non-mitogenic anti-CD3 to induce tolerance in T1D has had some initial success [6-8], but there are still concerns about efficacy, cytokine release syndrome, and viral reactivation. Attempts to improve safety via oral administration of anti-CD3 resulted in some positive immunoregulatory effects in a small study involving 15 healthy controls [9]. Larger patient studies will be needed to clearly define any clinical benefit of oral vs. intravenous anti-CD3 treatment.
Induction of tolerance using autoantigenic peptides chemically cross-linked to splenic leukocytes (Ag-ECDI-SP) or other cellular carriers using ethylene carbodiimide (ECDI) has been shown to be a safer tolerance therapy [10] and is significantly more effective for the prevention and treatment of autoimmunity in various animal models than therapies using soluble antigen or broad spectrum immunosuppressive drugs (reviewed in [11]). Induction of tolerance using Ag-ECDI-SP was first shown to be an effective treatment in the experimental autoimmune encephalomyelitis (EAE) model of multiple sclerosis (MS) [11-13]. The therapy has recently proven safe and has shown some efficacy in a magnetic resonance imaging-controlled phase I clinical trial in relapsing-remitting MS patients at the Center for Multiple Sclerosis, University of Hamburg, Germany [14]. In addition to EAE and MS, the efficacy of Ag-ECDI-SP has been demonstrated in other immune-mediated inflammatory conditions, including T1D [15, 16], allergy [17], and islet transplant rejection [18]. This review focuses on the use and mechanisms of tolerance induction using Ag-ECDI-SP and Ag-coupled biodegradable poly(lactide-co-glycolide) (Ag-PLG) nanoparticles in experimental mouse models of T1D.
2. Mechanism of Ag-ECDI-SP tolerance
The two signal hypothesis for CD4+ T cell activation requires both stimulation of the T cell receptor (TCR) (signal 1) and costimulatory molecules, e.g. CD28/CD80/CD86 (signal 2). Engagement of Ag/MHC by the TCR in the absence of costimulation results in T cell anergy or activation-induced death, and is a crucial mechanism for both central and peripheral tolerance [19, 20]. In vitro studies show Ag-EDCI-SP can directly engage TCR signaling in the absence of costimulation, resulting in long lasting T cell anergy [21, 22]. These unresponsive cells remain viable and in a non-proliferative state that can be reversed by the addition of interleukin 2 (IL-2) [21].
While there is clear evidence for the role of T cell anergy in Ag-ECDI-SP tolerance, subsequent studies have shown that splenocytes treated with antigen-processing inhibitors, MHC knock-out splenocytes, or donor red blood cells (RBCs) lacking MHC class II expression can still induce tolerance [16, 23, 24]. This suggests that T cell anergy through direct TCR engagement of the Ag-ECDI-SP is not required, rather tolerance induction is primarily achieved through indirect mechanisms involving host antigen-presenting cells (APCs) [23]. The exact mechanisms for indirect tolerance are unclear. However, it is hypothesized that Ag-ECDI-SP utilize processes that are inherently in place to maintain peripheral tolerance during normal tissue turnover. During natural cell turnover, clearance of apoptotic cells occurs in a pro-tolerogenic fashion. Apoptotic cells release immunosuppressive cytokines and alter the expression of their surface proteins [25, 26]. These changes are recognized by apoptotic cell-sensing receptors, resulting in engulfment by macrophages and other phagocytic cells. After engulfment, tolerance can be established via several distinct mechanisms, including suppression of inflammatory cytokine production, release of anti-inflammatory cytokines, and modulation of antigen presentation and costimulatory molecules by APCs [27].
Fixation of Ag on the surface of splenocytes by ECDI readily induces apoptotic cell death [23]. Similar to apoptotic cells produced during normal cell death, Ag-EDCI-SP can be phagocytized by macrophages and dendritic cells (DCs) [23, 24]. Trafficking studies show that apoptotic Ag-ECDI-SP traffic to the host spleen, accumulate in the splenic marginal zone, and are then internalized. Processed antigens are represented by resident APCs, including marginal zone macrophages (MZMs) and CD8+ DCs [27]. Splenectomy abrogates tolerance induction, suggesting that uptake and processing of Ag-ECDI-SP in a non-immunogenic fashion requires MZMs and/or splenic DCs [27]. Similar pathways have been cited for related tolerogenic approaches. Tolerance induced by infusion of UVB-treated NIT-1 cells, or the administration of DCs pulsed with apoptotic bodies from beta-cells, are thought to occur by utilizing the same underlying mechanisms [28, 29]. The exact mechanisms by which Ag-EDCI-SP promote tolerance induction still remain unclear. However, it is likely that several distinct mechanisms are working synergistically to increase therapeutic efficacy.
3. Ag-ECDI-SP tolerance in T1D
3.1 Insulin-ECDI-SP in NOD mice
The non-obese diabetic (NOD) mouse is the most widely used animal model for T1D and has been extensively characterized [30]. In contrast to other mouse models of autoimmune diseases, NOD mice develop spontaneous T1D similar to the human diabetes, in both the genetic and environmental factors that influence disease susceptibility, and in the nature of immune responses that mediate pathology [30-32]. Extensive studies using congenic mice have identified multiple insulin-dependent loci that confer susceptibility to T1D; many of these genes have homologous counterparts in human disease [33-35]. The majority of these genes play important roles in immune regulation, including the induction and maintenance of central and peripheral tolerance [36]. It should be noted that there are many differences between human T1D and the NOD disease, which may limit the translation of findings in this mouse model to the treatment of the human disease [30, 32, 37]. However, humans and NOD mice share many of the same antigenic targets and specific CD4+ and CD8+ epitopes found on insulin (INS), islet-specific G6Pase catalytic subunit-related protein (IGRP), heat shock protein 60 (HSP60), and islet cell antigen 512 (IA-2) [38].
Many lines of evidence show that insulin, specifically insulin B9-23 is the primary initiating epitope in T1D [39-41]. NOD mice with knockouts of both native insulin genes that express a mutated proinsulin transgene (tyrosine to alanine substitution at position B16 in preproinsulin) do not develop insulitis, lack islet autoantibodies, and are completely protected from diabetes. This demonstrates that immune recognition of the InsB9-23 epitope is required for initiating pathogenesis, depending on the tyrosine residue at position 16 [40]. In a study, tolerance induction by genetic overexpression of preproinsulin on APCs inhibited autoreactivity to insulin, but not IGRP, and prevented the onset of disease [41]. Taken together, these data show insulin to be a critical autoantigen in the early initiation and progressive pathology of T1D. Treatment with insulin-ECDI-SP induced long-term remission in up to 50% of new onset diabetic NOD mice [15]. These findings highlighted that one of the few therapies (both antigen-specific and non-specific) can reverse diabetes in NOD mice after clinical onset of disease.
Treatment of young NOD mice with insulin B9-23-ECDI-SP or insulin-ECDI-SP before onset protected up to 100% of mice from disease onset [16]. Administration of Insulin-ECDI-SP reduced the infiltration of immune cells in the pancreatic islets and blocked the induction of DTH responses in an antigen-specific manner. Insulin coupled to donor RBCs also effectively protected NOD mice from onset of T1D, demonstrating that tolerance induction primarily occured through indirect mechanisms [16]. Depletion of T regulatory cells (Tregs) at the time of treatment abolished the protective effects of Ag-ECDI-SP treatment, suggesting Tregs were crucial for efficient induction of tolerance. Strikingly, treatment with insulinB9-23-ECDI-SP was effective only when mice were treated in the early stages of disease (4-6 weeks); it was less effective when administered at later stages of the disease (19-21 weeks) [16]. This suggests that B9-23 is a dominant initiating epitope, but autoimmune responses to insulin epitopes distinct from InsB9-23 emerge during disease progression via epitope spreading. No protection was seen using other T cell epitopes that have been implicated in the pathogenesis of T1D, including GAD65206-220, GAD65217-236, GAD65509-528, GAD65524-543, IGRP206-214, and NRP-V7 [15, 16]. This suggests, while T cell reactivity to many islet antigens develops during the course of T1D, only a few antigens may be critical for inducing pathogenesis, and less dominant antigens may not be suitable targets for antigen-specific therapies.
3.2 p31-ECDI-SP in adoptive transfer of BDC2.5 TCR transgenic cells
Ag-ECDI-SP prevent autoimmunity in the BDC2.5 TCR transgenic (Tg) mouse model of T1D. BDC2.5 TCR Tg mice express a TCR that recognizes an epitope on a pancreatic β-cell antigen, chromogranin A (ChgA) [42]. Recent work has shown that this TCR Tg specifically targets an epitope found on the chromogranin A-derived peptide vasostatin-1 (ChgA29-42) [43]. The BDC2.5 TCR Tg mouse was originally derived from a highly pathogenic CD4+ T cell clone isolated from the pancreas of a diabetic NOD mouse, and has been used extensively to study the autoimmune response in T1D [44].
Adoptive transfer of BDC2.5 Tg T cells activated in vitro with 1040-p31 peptide (p31), a mimetope of ChgA29-42, induces a highly aggressive autoimmune response in which 100% of recipient mice develop T1D within 5-7 days. Treatment with p31-ECDI-SP reduced the levels of the mononuclear infiltrate in pancreatic islets, and protected mice from T1D [15]. Tolerized BDC2.5 T cells had reduced proliferative responses and reduced interferon γ (IFN-γ) and IL-2 production. CD4+ T cells from p31-ECDI-SP-treated mice upregulate cytotoxic T lymphocyte antigen 4 (CTLA4) and programmed cell death 1 (PD-1), an inhibitory member of the CD28 family of costimulatory receptors. Blockade of CTLA-4 or the PD-1/PD-L1 pathway abrogates tolerance induction in the BDC2.5 AT model for T1D. However, in tolerized mice, treatment with anti-PDL-1, but not anti-CTLA-4, reverses T cell anergy resulting in diabetes. This demonstrates that the PD-1/PD-L1 inhibitory pathway is required for both induction and long-term maintenance of tolerance. No differences in Treg populations were reported in p31-ECDI-SP-tolerized mice, and protection was maintained in recipients of Treg-depleted BDC2.5 CD4 T cells [15]. In contrast to insulin-ECDI-SP, in spontaneous T1D and other models of autoimmunity, these data indicate that p31-ECDI-SP in the BDC2.5 AT model occur in a Treg-independent manner, and tolerance is established via T cell-intrinsic mechanisms.
3.3 Ag-ECDI-SP in "humanized" NOD.β2mnull.HHD model of T1D
Efficacy of Ag-ECDI-SP has also been demonstrated in a humanized NOD mouse model. NOD.β2mnull.HHD mice lack murine-derived MHC I and instead transgenically express human HLA-A2.1 molecules [24]. Genetic studies have linked HLA-A.2.1 to T1D susceptibility in humans, and Tg expression of the HLA haplotype in NOD mice accelerates the onset of disease [45]. NOD.β2mnull.HHD have been shown to present IGRP (IGRP228-236, IGRP265-273, and IGRP337-345) and insulin (INS1 L3-11, INS1 B5-14, and INS1/2 A2-10) to activate CD8+ T cells [45-47]. Homologous human peptides for Ins A2-10, Ins B5-15, IGRP228-236, and IGRP265-273 can also activate NOD.β2mnull.HHD CD8+ T cells [46, 47]. This striking overlap in the HLA-A2.1-restricted CD8+ responses in NOD and human T1D provides a powerful model system to examine the viability of Ag-ECDI-SP-induced tolerance in a more clinically relevant animal model system.
NOD.β2mnull.HHD mice treated with Ag-ECDI-SP coupled with a combination of IGRP228-236 and IGRP265-273 peptides blocked IFN-γ production in NOD.β2mnull.HHD CD8+ T cells, and protected mice from the onset of T1D [24]. Protective effects required tolerance induction to both IGRP peptides as no differences were seen in mice treated with IGRP228-236 or IGRP265-273 alone. Tolerance was effectively established using MHC class I-deficient donor cells, and ECDI-fixed cells were rapidly taken up by host splenic DCs, similar to previous studies using CD4+ T cell-restricted autoantigens presented by APCs [27]. This indicates that CD8+ T cell tolerance induction occurs primarily through indirect mechanisms via uptake and processing of Ag-ECDI-SP by host APCs. This work demonstrates potent tolerance induction to diabetogenic CD8+ T epitopes. The efficacy of Ag-ECDI-SP to induce tolerance to CD4+ T cell epitopes, using a combination of multiple peptides, has previously been demonstrated in EAE [48]. Most importantly, effective tolerance in the humanized NOD.β2mnull.HHD mouse model supports the potential of Ag-ECDI-SP-induced tolerance in human disease, and provides a model system to test other potential antigenic targets that are important for human pathogenesis to facilitate efforts in clinical translation of this therapy.
3.4 Ag-ECDI-SP in islet transplantation
Preclinical studies demonstrate that Ag-ECDI-SP represent a potent therapy for T1D, especially when administered before the onset of hyperglycemia. As it is possible to select pre-T1D patients with some certainty based on family history of T1D, HLA typing, and the development of autoantibodies to β-cell antigens, one could envision the effective use of patient peripheral blood mononuclear cells (PBMCs), fixed with diabetogenic antigens using ECDI, as a preventative therapy for T1D. At diagnosis, the patients have usually lost 80-95% of their β-cell mass, which continues to decline after initiation of insulin replacement therapy.
Islet cell transplantation can restore endogenous insulin production and reduce or eliminate the need for insulin replacement therapy. Recent improvements in sourcing, harvesting, and transplantation of islet cells, as well as advancements in post-transplant immunosuppressive therapy, have dramatically improved the outcomes in ongoing clinical trials [49, 50]. While islet transplantation can replenish β-cells and restore insulin production, it does not address the underlying autoimmunity to islet antigens. There is still a substantial risk for eventual rejection of the transplanted islets and the development of opportunistic infections due to long-term maintenance immunosuppression. The hope is to utilize autologous patient Ag-ECDI-PBMCs to re-establish tolerance to β-cell antigens in combination with induction of tolerance to alloantigens present on the islet cell donor. Indefinite tolerance to subsequent allogeneic islet cell transplantation was established in diabetic mice by i.v. infusion of donor splenic leukocytes rendered apoptotic by fixation using ECDI, with no requirement for maintenance immunosuppression [18]. Treated mice exhibited reduced DTH responses to donor antigens, were devoid of donor-specific antibodies (Abs), and showed reduced donor-specific proliferation and IFN-γ production in mixed lymphocyte cultures.
Similar to what was observed in Ag-ECDI-SP-induced tolerance in autoimmune diabetes, PD-1/PD-L1 signaling and Tregs are crucial for the efficient induction of tolerance to alloantigens. In mice treated with ECDI-fixed donor leukocytes, accumulation of Tregs is seen in peri-islet-infiltrating cells in islet grafts [51, 52]. Depletion of Tregs or treatment with anti-TGF-β, which has been shown to be effective in the induction of Tregs in vivo prior to or at the time treatment, abolished the protective effects of ECDI-fixed donor leukocytes [18]. However, if depletion of Tregs was induced after tolerance no graft rejection occurred, suggesting that Tregs are required for induction of tolerance, but that maintenance of tolerance was probably mediated through T cell-intrinsic tolerogenic mechanisms.
3.5 Ag-ECDI-PLGA nanoparticles induce tolerance
The studies discussed above demonstrate the efficacy of protein or peptide antigens coupled to splenic or peripheral blood leukocytes to induce antigen-specific tolerance and thus to prevent or reverse T1D. However, the technical complexity and costs associated with sourcing and peptide coupling of donor cells under good manufacturing practice (GMP) procedures may likely limit widespread clinical translation of this technique.
To overcome these potential challenges, inert polystyrene beads (PSB) or biodegradable poly(lactic-co-glycolic acid) (PLGA) nanoparticles are being explored as surrogates for apoptotic debris to serve as substitutes for cellular vehicles. PLGA has been approved for therapeutic use by the US Food and Drug Administration and overcomes stability, storage, and manufacturing issues associated with using the apoptotic cell-based strategy [53]. PLGA particles are highly customizable; for example, PLGA particles can be carboxylated, making them suitable for covalent cross-linking of target peptides via ECDI. Recent work has shown the efficacy of encephalitogenic myelin epitopes coupled to PSB and PLGA nanoparticles to both prevent and treat autoimmunity in the EAE mouse model of MS [54].
Similar to the use of Ag-ECDI-SP, efficient induction of tolerance using PSB induces both T cell anergy and the activation of antigen-specific Treg cells. The therapeutic effects of Ag-ECDI-PSB require the expression of the scavenger macrophage receptor with collagenous structure (MARCO). MARCO is predominantly expressed by phagocytes, and is implicated in the entry and clearance of particulate debris [55, 56]. This suggests that inert nanoparticles travel to tolerogenic APCs where they enter the cells via the MARCO scavenger receptor similar to apoptotic carrier cells, and trigger similar mechanisms to induce antigen-specific tolerance (Figure 1). Thus, PSB and PLGA nanoparticles provide a potent platform to couple autoantigens and possibly donor MHC antigens for induction of tolerance, which need to be evaluated for safety and efficacy in appropriate clinical trials. Preliminary experiments show that tolerance can be achieved by this method. Infusion of Ag-ECDI-PLGA nanoparticles in the BDC2.5 adoptive transfer model of T1D prevents the onset of hyperglycemia mediated by transfer of activated transgenic T cells to NOD.SCID recipients (manuscript in preparation). Significantly reduced numbers of BDC2.5 T cells travel to the pancreas of mice treated with p31-ECDI-PLGA nanoparticles. Those T cells show a significantly reduced activity regarding the production of the pro-inflammatory cytokines, IFN-γ and TNF-α. Ongoing studies aim to determine the exact mechanism for Ag-ECDI-PLGA tolerance induction in the BDC2.5 AT transfer model of T1D, and to compare and contrast these mechanisms with those that have been described in tolerance induction using apoptotic cell-based therapy.
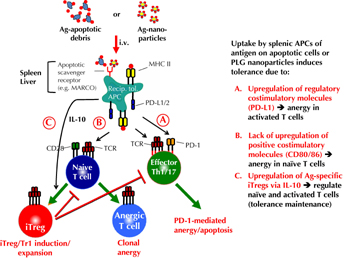 |
 |
Figure 1. Proposed mechanisms of tolerance induced by antigen-coupled apoptotic cells and Ag-coupled PLG nanoparticles. Intravenous infusion of Ag-coupled ECDI-fixed apoptotic cells or Ag-coupled biodegradable PLG nanoparticles enter 'tolerogenic' APCs in the spleen and liver via scavenger receptors, e.g. MARCO [27, 54]. Uptake leads to presentation of the coupled Ag by MHC molecules on the host splenic and liver APCs. Concomitantly, signals resulting from the scavenger receptor uptake cause upregulation of regulatory costimulatory molecules, e.g. PD-L1/2 without upregulation of positive costimulatory molecules, e.g. CD80 and CD86, as well as the production of IL-10 and TGF-β [27]. A. Host tolerogenic APCs induce anergy/apoptosis in activated antigen-specific T cells via engagement of PD-1 which is upregulated on activated T cells [27, 60-62]. B. Host tolerogenic APCs induce anergy in naive antigen-specific T cells by presenting cognate Ag/MHC in the absence of positive costimulatory interactions (i.e. signal one in the absence of signal 2) [21]. C. PD-L1 expressing, IL-10-producing host tolerogenic APCs induce the induction/expansion of antigen-specific induced regulatory T cells (Tregs)/Tr1 cells, which synergistically act to regulate the differentiation of naive T cells to become pathogenic effector T cells, and which regulate the effector function/trafficking of activated pathogenic T cells [27, 60, 63, 64]. |
|
PLGA nanoparticles can be manufactured to allow autoantigens to be coupled to the particle surface, or to allow encapsulation of antigen or other tolerance-promoting substances in nanospheres, enabling controlled sustained release and more efficient induction of tolerance [57]. Indeed, our recent results show that PLP139-151 encapsulated within carboxylated PLGA nanospheres can induce specific tolerance, resulting in inhibition of relapsing EAE. Current studies aim to explore whether the expression of various ligands on the surface of Ag-ECDI-PLGA nanoparticles can result in a more targeted uptake by tolerogenic APCs, and whether the encapsulation of regulatory/inhibitory cytokines, such as IL-10 and/or TGF-β, will more effectively induce antigen-specific tolerance.
4. Perspectives
Ag-ECDI-SP can be used to prevent and/or treat autoimmunity in multiple mouse models of T1D. Ag-ECDI peripheral blood leucocyte (PBL) therapy shows promising results in early clinical trials on MS. A clinical trial for the prevention of T1D is currently being considered by the Immune Tolerance Network (ITN).
NOD mice have served as a powerful model for the identification of pathogenic autoantigens in T1D pathogenesis. However, the relative contribution of these autoantigens in human disease is not clear. For example, mice have reduced levels of GAD65 in the pancreatic islets compared to rat or human islets, suggesting that GAD maybe be a more critical antigen in human disease [58, 59]. Successful clinical translation of tolerance therapies will require the identification of autoantigen(s) relevant for the pathogenesis of human T1D. Tolerance induction using cell-based therapies employing Ag-ECDI-SP may be hampered by cost and complexity issues. The use of biodegradable PLGA nanoparticles provides a more stable donor vehicle which can be readily customized and easily manufactured under GMP conditions.
Autoantigen-specific tolerance would avoid side effects associated with current immunomodulatory agents, and would ideally be applied to patients at high risk of T1D prior to the onset of overt disease. Tolerogenic intervention at this early stage of disease would likely require the targeting of fewer potential diabetogenic antigens as epitope spreading would not be advanced at this point, and would be more likely to result in permanent regeneration/restoration of β-cell function. Reversal of long-standing diabetes in patients who have lost the vast majority of their β-cells would require a combined approach, including tolerance to autoantigens for the regulation of autoimmunity, which initially destroyed the β-cells, and tolerance to alloantigens on donor islets used to replace insulin production. Autoantigen-specific tolerance could be induced by Ag-coupled autologous PBMCs (Ag-ECDI-PBMCs) [16] or Ag-coupled PLGA nanoparticles [54] (manuscript in preparation). Tolerance to donor alloantigens can be readily induced by i.v. infusion of ECDI-fixed donor leukocytes administered seven days prior to and one day after allogeneic islet transplantation [18]. Since tolerance induction with Ag-ECDI-SP can be achieved using apoptotic allogeneic leukocytes as autoantigen carriers [23], it may be possible to use allogeneic Ag-ECDI leukocytes to simultaneously induce tolerance to autoantigens and alloantigens.
Our recent experiments have shown that i.v. infusion of apoptotic ECDI-fixed xenogeneic rat or pig leukocytes into diabetic C57Bl/6 mice can induce indefinite xenoantigen-specific tolerance when combined with B cell depletion, using anti-CD20 along with a short course of rapamycin (manuscript in revision). Translation of xenogeneic tolerance to T1D patients would be an important advance as the ability to use porcine islets to reverse T1D would provide a more consistent and readily available source of insulin-producing β-cells than the current use of islets purified from non-MHC-matched cadaver donors.
Disclosure: The authors report no conflict of interests.
Acknowledgments:
This work was supported by NIH grant NS026543 and EB013198, and Juvenile Diabetes Research Foundation grant 17-2011-343.
References
- Bluestone JA, Herold K, Eisenbarth G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature 2010. 464:1293-1300. [DOD] [CrossRef]
- Ludvigsson J, Krisky D, Casas R, Battelino T, Castano L, Greening J, Kordonouri O, Otonkoski T, Pozzilli P, Robert JJ, et al. GAD65 antigen therapy in recently diagnosed type 1 diabetes mellitus. New Engl J Med 2012. 366:433-442. [DOD] [CrossRef]
- Chen Y, Kuchroo VK, Inobe J, Hafler DA, Weiner HL. Regulatory T cell clones induced by oral tolerance: suppression of autoimmune encephalomyelitis. Science 1994. 265:1237-1240. [DOD] [CrossRef]
- Luo X, Herold KC, Miller SD. Immunotherapy of type 1 diabetes: where are we and where should we be going? Immunity 2010. 32:488-499. [DOD]
- Sherr J, Sosenko J, Skyler JS, Herold KC. Prevention of type 1 diabetes: the time has come. Nat Clin Pract Endocrinol Metab 2008. 4:334-343. [DOD]
- Herold KC, Gitelman SE, Masharani U, Hagopian W, Bisikirska B, Donaldson D, Rother K, Diamond B, Harlan DM, Bluestone JA. A single course of anti-CD3 monoclonal antibody hOKT3gamma1(Ala-Ala) results in improvement in C-peptide responses and clinical parameters for at least 2 years after onset of type 1 diabetes. Diabetes 2005. 54:1763-1769. [DOD] [CrossRef]
- Keymeulen B, Vandemeulebroucke E, Ziegler AG, Mathieu C, Kaufman L, Hale G, Gorus F, Goldman M, Walter M, Candon S, et al. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med 2005. 352:2598-2608. [DOD] [CrossRef]
- Herold KC, Gitelman S, Greenbaum C, Puck J, Hagopian W, Gottlieb P, Sayre P, Bianchine P, Wong E, Seyfert-Margolis V, et al. Treatment of patients with new onset Type 1 diabetes with a single course of anti-CD3 mAb Teplizumab preserves insulin production for up to 5 years. Clin Immunol 2009. 132:166-173. [DOD] [CrossRef]
- Ilan Y, Zigmond E, Lalazar G, Dembinsky A, Ben Ya'acov A, Hemed N, Kasis I, Axelrod E, Zolotarov L, Klein A, et al. Oral administration of OKT3 monoclonal antibody to human subjects induces a dose-dependent immunologic effect in T cells and dendritic cells. J Clin Immunol 2010. 30:167-177. [DOD] [CrossRef]
- Smith CE, Eagar TN, Strominger JL, Miller SD. Differential induction of IgE-mediated anaphylaxis after soluble vs. cell-bound tolerogenic peptide therapy of autoimmune encephalomyelitis. Proc Natl Acad Sci USA 2005. 102:9595-9600. [DOD] [CrossRef]
- Miller SD, Turley DM, Podojil JR. Antigen-specific tolerance strategies for the prevention and treatment of autoimmune disease. Nat Rev Immunol 2007. 7:665-677. [DOD] [CrossRef]
- Kennedy MK, Tan LJ, Dal Canto MC, Tuohy VK, Lu ZJ, Trotter JL, Miller SD. Inhibition of murine relapsing experimental autoimmune encephalomyelitis by immune tolerance to proteolipid protein and its encephalitogenic peptides. J Immunol 1990. 144:909-915. [DOD]
- Tan LJ, Kennedy MK, Miller SD. Regulation of the effector stages of experimental autoimmune encephalomyelitis via neuroantigen-specific tolerance induction. II. Fine specificity of effector T cell inhibition. J Immunol 1992. 148:2748-2755. [DOD]
- Lutterotti A, Yusef S, Sputtek A, Sturner K, Stellmann JP, Breiden P, Reinhardt S, Schulze C, Bester M, Heesen C, et al. Induction of tolerance by autologous peptide coupled cells in multiple sclerosis. Sci Transl Med 2013. In press. [DOD]
- Fife BT, Guleria I, Gubbels Bupp M, Eagar TN, Tang Q, Bour-Jordan H, Yagita H, Azuma M, Sayegh MH, Bluestone JA. Insulin-induced remission in new-onset NOD mice is maintained by the PD-1-PD-L1 pathway. J Exp Med 2006. 203:2737-2747. [DOD] [CrossRef]
- Prasad S, Kohm AP, McMahon JS, Luo X, Miller SD. Pathogenesis of NOD diabetes is initiated by reactivity to the insulin B chain 9-23 epitope and involves functional epitope spreading. J Autoimm 2012. 39:347-353. [DOD] [CrossRef]
- Smarr CB, Hsu CL, Byrne AJ, Miller SD, Bryce PJ. Antigen-fixed leukocytes tolerize Th2 responses in mouse models of allergy. J Immunol 2011. 187:5090-5098. [DOD] [CrossRef]
- Luo X, Pothoven KL, McCarthy D, DeGutes M, Martin A, Getts DR, Xia G, He J, Zhang X, Kaufman DB, et al. ECDI-fixed allogeneic splenocytes induce donor-specific tolerance for long-term survival of islet transplants via two distinct mechanisms. Proc Natl Acad Sci USA 2008. 105:14527-14532. [DOD] [CrossRef]
- Sharpe AH. Mechanisms of costimulation. Immunol Rev 2009. 229:5-11. [DOD] [CrossRef]
- Podojil JR, Miller SD. Molecular mechanisms of T-cell receptor and costimulatory molecule ligation/blockade in autoimmune disease therapy. Immunol Rev 2009. 229:337-355. [DOD] [CrossRef]
- Jenkins MK, Schwartz RH. Antigen presentation by chemically modified splenocytes induces antigen-specific T cell unresponsiveness in vitro and in vivo. J Exp Med 1987. 165:302-319. [DOD]
- Eagar TN, Karandikar NJ, Bluestone J, Miller SD. The role of CTLA-4 in induction and maintenance of peripheral T cell tolerance. Eur J Immunol 2002. 32:972-981. [DOD] [CrossRef]
- Turley DM, Miller SD. Peripheral tolerance Induction using ethylenecarbodiimide-fixed APCs uses both direct and indirect mechanisms of antigen presentation for prevention of experimental autoimmune encephalomyelitis. J Immunol 2007. 178:2212-2220. [DOD]
- Niens M, Grier AE, Marron M, Kay TW, Greiner DL, Serreze DV. Prevention of "humanized" diabetogenic CD8 T-cell responses in HLA-transgenic NOD mice by a multipeptide coupled-cell approach. Diabetes 2011. 60:1229-1236. [DOD] [CrossRef]
- Griffith TS, Ferguson TA. Cell death in the maintenance and abrogation of tolerance: the five Ws of dying cells. Immunity 2011. 35:456-466. [DOD] [CrossRef]
- A-Gonzalez N, Bensinger SJ, Hong C, Beceiro S, Bradley MN, Zelcer N, Deniz J, Ramirez C, Diaz M, Gallardo G, et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity 2009. 31(2):245-258. [DOD] [CrossRef]
- Getts DR, Turley DM, Smith CE, Harp CT, McCarthy D, Feeney EM, Getts MT, Martin AJ, Luo X, Terry RL, et al. Tolerance induced by apoptotic antigen-coupled leukocytes is induced by PD-L1+ and IL-10-producing splenic macrophages and maintained by T regulatory cells. J Immunol 2011. 187:2405-2417. [DOD] [CrossRef]
- Xia CQ, Peng R, Qiu Y, Annamalai M, Gordon D, Clare-Salzler MJ. Transfusion of apoptotic beta-cells induces immune tolerance to beta-cell antigens and prevents type 1 diabetes in NOD mice. Diabetes 2007. 56:2116-2123. [DOD] [CrossRef]
- Marin-Gallen S, Clemente-Casares X, Planas R, Pujol-Autonell I, Carrascal J, Carrillo J, Ampudia R, Verdaguer J, Pujol-Borrell R, Borras FE, et al. Dendritic cells pulsed with antigen-specific apoptotic bodies prevent experimental type 1 diabetes. Clin Exp Immunol 2010. 160:207-214. [DOD] [CrossRef]
- von Herrath M, Nepom GT. Animal models of human type 1 diabetes. Nat Immunol 2009. 10:129-132. [DOD] [CrossRef]
- Driver JP, Chen YG, Zhang W, Asrat S, Serreze DV. Unmasking genes in a type 1 diabetes-resistant mouse strain that enhances pathogenic CD8 T-cell responses. Diabetes 2011. 60:1354-1359. [DOD] [CrossRef]
- von Herrath M, Nepom GT. Remodeling rodent models to mimic human type 1 diabetes. Eur J Immunol 2009. 39:2049-2054. [DOD] [CrossRef]
- Maier LM, Wicker LS. Genetic susceptibility to type 1 diabetes. Curr Opin Immunol 2005. 17:601-608. [DOD] [CrossRef]
- Wicker LS, Clark J, Fraser HI, Garner VE, Gonzalez-Munoz A, Healy B, Howlett S, Hunter K, Rainbow D, Rosa RL, et al. Type 1 diabetes genes and pathways shared by humans and NOD mice. J Autoimmun 2005. 25(Suppl):29-33. [DOD] [CrossRef]
- Todd JA. Etiology of type 1 diabetes. Immunity 2010. 32:457-467. [DOD] [CrossRef]
- Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol 2005. 23:447-485. [DOD] [CrossRef]
- Kachapati K, Adams D, Bednar K, Ridgway WM. The non-obese diabetic (NOD) mouse as a model of human type 1 diabetes. Methods Mol Biol 2012. 933:3-16. [DOD]
- DiLorenzo TP. Multiple antigens versus single major antigen in type 1 diabetes: arguing for multiple antigens. Diabetes Metab Res Rev 2011. 27:778-783. [DOD] [CrossRef]
- Moriyama H, Abiru N, Paronen J, Sikora K, Liu E, Miao D, Devendra D, Beilke J, Gianani R, Gill RG, et al. Evidence for a primary islet autoantigen (preproinsulin 1) for insulitis and diabetes in the nonobese diabetic mouse. Proc Natl Acad Sci USA 2003. 100:10376-10381. [DOD] [CrossRef]
- Nakayama M, Abiru N, Moriyama H, Babaya N, Liu E, Miao DM, Yu LP, Wegmann DR, Hutton JC, Elliott JF, et al. Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice. Nature 2005. 435:220-223. [DOD] [CrossRef]
- Krishnamurthy B, Dudek NL, McKenzie MD, Purcell AW, Brooks AG, Gellert S, Colman PG, Harrison LC, Lew AM, Thomas HE, et al. Responses against islet antigens in NOD mice are prevented by tolerance to proinsulin but not IGRP. J Clin Invest 2006. 116:3258-3265. [DOD] [CrossRef]
- Stadinski BD, Delong T, Reisdorph N, Reisdorph R, Powell RL, Armstrong M, Piganelli JD, Barbour G, Bradley B, Crawford F, et al. Chromogranin A is an autoantigen in type 1 diabetes. Nat Immunol 2010. 11:225-231. [DOD] [CrossRef]
- Nikoopour E, Sandrock C, Huszarik K, Krougly O, Lee-Chan E, Masteller EL, Bluestone JA, Singh B. Cutting Edge: Vasostatin-1-derived peptide ChgA29-42 is an antigenic epitope of diabetogenic BDC2.5 T cells in nonobese diabetic mice. J Immunol 2011. 186:3831-3835. [DOD] [CrossRef]
- Katz JD, Wang B, Haskins K, Benoist C, Mathis D. Following a diabetogenic T cell from genesis through pathogenesis. Cell 1993. 74:1089-1100. [DOD] [CrossRef]
- Serreze DV, Marron MP, Dilorenzo TP. "Humanized" HLA transgenic NOD mice to identify pancreatic beta cell autoantigens of potential clinical relevance to type 1 diabetes. Ann N Y Acad Sci 2007. 1103:103-111. [DOD] [CrossRef]
- Takaki T, Marron MP, Mathews CE, Guttmann ST, Bottino R, Trucco M, DiLorenzo TP, Serreze DV. HLA-A*0201-restricted T cells from humanized NOD mice recognize autoantigens of potential clinical relevance to type 1 diabetes. J Immunol 2006. 176:3257-3265. [DOD]
- Jarchum I, Baker JC, Yamada T, Takaki T, Marron MP, Serreze DV, DiLorenzo TP. In vivo cytotoxicity of insulin-specific CD8+ T-cells in HLA-A*0201 transgenic NOD mice. Diabetes 2007. 56:2551-2560. [DOD] [CrossRef]
- Smith CE, Miller SD. Multi-peptide coupled-cell tolerance ameliorates ongoing relapsing EAE associated with multiple pathogenic autoreactivities. J Autoimmun 2006. 27:218-231. [DOD] [CrossRef]
- Shapiro AM, Ricordi C, Hering BJ, Auchincloss H, Lindblad R, Robertson RP, Secchi A, Brendel MD, Berney T, Brennan DC, et al. International trial of the Edmonton protocol for islet transplantation. N Engl J Med 2006. 355:1318-1330. [DOD] [CrossRef]
- Barton FB, Rickels MR, Alejandro R, Hering BJ, Wease S, Naziruddin B, Oberholzer J, Odorico JS, Garfinkel MR, Levy M, et al. Improvement in outcomes of clinical islet transplantation: 1999-2010. Diabetes Care 2012. 35:1436-1445. [DOD] [CrossRef]
- Kheradmand T, Wang S, Gibly RF, Zhang X, Holland S, Tasch J, Graham JG, Kaufman DB, Miller SD, Shea LD, et al. Permanent protection of PLG scaffold transplanted allogeneic islet grafts in diabetic mice treated with ECDI-fixed donor splenocyte infusions. Biomaterials 2011. 32:4517-4524. [DOD] [CrossRef]
- Kheradmand T, Wang S, Bryant J, Tasch JJ, Lerret N, Pothoven KL, Houlihan JL, Miller SD, Zhang ZJ, Luo X. Ethylenecarbodiimide-fixed donor splenocyte infusions differentially target direct and indirect pathways of allorecognition for induction of transplant tolerance. J Immunol 2012. 189:804-812. [DOD] [CrossRef]
- Holgado MA, Alvarez-Fuentes J, Fernandez-Arevalo M, Arias JL. Possibilities of poly(D,L-lactide-co-glycolide) in the formulation of nanomedicines against cancer. Curr Drug Targets 2011. 12:1096-1111. [DOD] [CrossRef]
- Getts DR, Martin AJ, McCarthy DP, Terry RL, Hunter ZN, Yap WT, Getts MT, Pleiss M, Luo X, King NJ, et al. Microparticles bearing encephalitogenic peptides induce T-cell tolerance and ameliorate experimental autoimmune encephalomyelitis. Nat Biotechnol 2012. 30:1217-1224. [DOD] [CrossRef]
- Kanno S, Furuyama A, Hirano S. A murine scavenger receptor MARCO recognizes polystyrene nanoparticles. Toxicol Sci 2007. 97:398-406. [DOD] [CrossRef]
- Thakur SA, Hamilton R Jr, Pikkarainen T, Holian A. Differential binding of inorganic particles to MARCO. Toxicol Sci 2009. 107:238-246. [DOD] [CrossRef]
- Phillips B, Nylander K, Harnaha J, Machen J, Lakomy R, Styche A, Gillis K, Brown L, Lafreniere D, Gallo M, et al. A microsphere-based vaccine prevents and reverses new-onset autoimmune diabetes. Diabetes 2008. 57:1544-1555. [DOD] [CrossRef]
- Kim J, Richter W, Aanstoot HJ, Shi Y, Fu Q, Rajotte R, Warnock G, Baekkeskov S. Differential expression of GAD65 and GAD67 in human, rat, and mouse pancreatic islets. Diabetes 1993. 42:1799-1808. [DOD] [CrossRef]
- Burton AR, Baquet Z, Eisenbarth GS, Tisch R, Smeyne R, Workman CJ, Vignali DA. Central nervous system destruction mediated by glutamic acid decarboxylase-specific CD4+ T cells. J Immunol 2010. 184:4863-4870. [DOD] [CrossRef]
- Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, Sharpe AH. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med 2009. 206:3015-3029. [DOD] [CrossRef]
- Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev 2010. 236:219-242. [DOD] [CrossRef]
- Yadav D, Sarvetnick N. Costimulation and pancreatic autoimmunity: the PD-1/PD-L conundrum. Rev Diabet Stud 2006. 3:6-10. [DOD] [CrossRef]
- Roncarolo MG, Gregori S, Battaglia M, Bacchetta R, Fleischhauer K, Levings MK. Interleukin-10-secreting type 1 regulatory T cells in rodents and humans. Immunol Rev 2006. 212:28-50. [DOD] [CrossRef]
- Zhang H, Podojil JR, Luo X, Miller SD. Intrinsic and induced regulation of the age-associated onset of spontaneous experimental autoimmune encephalomyelitis. J Immunol 2008. 181:4638-4647. [DOD]
This article has been cited by other articles:

|
Controlled Delivery of Single or Multiple Antigens in Tolerogenic Nanoparticles Using Peptide-Polymer Bioconjugates
Pearson RM, Casey LM, Hughes KR, Wang LZ, North MG, Getts DR, Miller SD, Shea LD
Mol Ther 2017. In press
|
|

|
Multicomponent Injectable Hydrogels for Antigen-Specific Tolerogenic Immune Modulation
Verbeke CS, Gordo S, Schubert DA, Lewin SA, Desai RM, Dobbins J, Wucherpfennig KW, Mooney DJ
Adv Healthc Mater 2017. In press
|
|

|
Bioengineering strategies for inducing tolerance in autoimmune diabetes
Baekkeskov S, Hubbell JA, Phelps EA
Adv Drug Deliv Rev 2017. In press
|
|
|
Regulatory T cell-based therapies for autoimmunity
Arellano B, Graber DJ, Sentman CL
Discov Med 2016. 22(119):73-80
|
|

|
An antigen-encapsulating nanoparticle platform for TH1/17 immune tolerance therapy
McCarthy DP, Yap JW, Harp CT, Song WK, Chen J, Pearson RM, Miller SD, Shea LD
Nanomedicine 2016. In press
|
|

|
Concise Review: Cell-Based Therapies and Other Non-Traditional Approaches for Type 1 Diabetes
Creusot RJ, Battaglia M, Roncarolo MG, Fathman CG
Stem Cells 2016. 34(4):809-819
|
|

|
Autoimmune Diabetes: An Overview of Experimental Models and Novel Therapeutics
You S, Chatenoud L
Methods Mol Biol 2016. 1371:117-42
|
|

|
TGF-beta1 along with other platelet contents augments Treg cells to suppress anti-FVIII immune responses in hemophilia A mice
Haribhai D, Luo X, Chen J, Jia S, Shi L, Schroeder JA, Weiler H, Aster RH, Hessner MJ, Hu J, Williams CB, Shi Q
Blood Adv 2016. 1(2):139-151
|
|
|
Immune Tolerance for Autoimmune Disease and Cell Transplantation
Luo X, Miller SD, Shea LD
Annu Rev Biomed Eng 2016. 18:181-205
|
|

|
Reversal of New-Onset Type 1 Diabetes With an Agonistic TLR4/MD-2 Monoclonal Antibody
Bednar KJ, Tsukamoto H, Kachapati K, Ohta S, Wu Y, Katz JD, Ascherman DP, Ridgway WM
Diabetes 2015. 64(10):3614-3626
|
|

|
Toward beta cell replacement for diabetes
Johannesson B, Sui L, Freytes DO, Creusot RJ, Egli D
EMBO J 2015. 34(7):841-855
|
|

|
Taming the TCR: antigen-specific immunotherapeutic agents for autoimmune diseases
Sauer EL, Cloake NC, Greer JM
Int Rev Immunol 2015. 34(6):460-485
|
|

|
Therapeutic applications of nanomedicine in autoimmune diseases: From immunosuppression to tolerance induction
Gharagozloo M, Majewski S, Foldvari M
Nanomedicine 2015. 11(4):1003-1018
|
|
|


)

