Chapter I. Pathogenesis
| Rev Diabet Stud,
2012,
9(4):148-168 |
DOI 10.1900/RDS.2012.9.148 |
Pathogenic Mechanisms in Type 1 Diabetes: The Islet is Both Target and Driver of Disease
Kate L. Graham1, Robyn M. Sutherland2,3, Stuart I. Mannering1,4, Yuxing Zhao1, Jonathan Chee1,4, Balasubramanian Krishnamurthy1,4, Helen E. Thomas1,4, Andrew M. Lew2,3, Thomas W.H. Kay1,4
1St. Vincent´s Institute of Medical Research, Fitzroy, Victoria, Australia
2The Walter and Eliza Hall Institute of Medical Research, Parkville, Victoria, Australia
3Department of Medical Biology, The University of Melbourne, Victoria, Australia
4Department of Medicine, The University of Melbourne, St. Vincent´s Hospital, Fitzroy, Victoria Australia
Address correspondence to: Thomas Kay, St Vincent's Institute, 41 Victoria Parade, Fitzroy, VIC, 3065, Australia, e-mail tkay@svi.edu.au
Manuscript submitted December 28, 2012; accepted January 22, 2013.
Keywords: type 1 diabetes, beta-cell, CTL, NOD mouse, islet, CD4+ T cell, insulitis, effector mechanism
Abstract
Recent advances in our understanding of the pathogenesis of type 1 diabetes have occurred in all steps of the disease. This review outlines the pathogenic mechanisms utilized by the immune system to mediate destruction of the pancreatic beta-cells. The autoimmune response against beta-cells appears to begin in the pancreatic lymph node where T cells, which have escaped negative selection in the thymus, first meet beta-cell antigens presented by dendritic cells. Proinsulin is an important antigen in early diabetes. T cells migrate to the islets via the circulation and establish insulitis initially around the islets. T cells within insulitis are specific for islet antigens rather than bystanders. Pathogenic CD4+ T cells may recognize peptides from proinsulin which are produced locally within the islet. CD8+ T cells differentiate into effector T cells in islets and then kill beta-cells, primarily via the perforin-granzyme pathway. Cytokines do not appear to be important cytotoxic molecules in vivo. Maturation of the immune response within the islet is now understood to contribute to diabetes, and highlights the islet as both driver and target of the disease. The majority of our knowledge of these pathogenic processes is derived from the NOD mouse model, although some processes are mirrored in the human disease. However, more work is required to translate the data from the NOD mouse to our understanding of human diabetes pathogenesis. New technology, especially MHC tetramers and modern imaging, will enhance our understanding of the pathogenic mechanisms.
Abbreviations: CCR7 – C-C motif chemokine receptor 7; CFSE – carboxyfluorescein succinimidyl ester; CTL – cytotoxic T lymphocyte; CXCL9 – C-X-C motif chemokine ligand 9; CXCR3 – C-X-C motif chemokine receptor 3; DC – dendritic cell; ELIspot – enzyme-linked immuno spot; FADD – Fas-associated death domain; GAD65 – glutamic acid decarboxylase 65; GFAP – glial fibrillary acidic protein; HLA – human leukocyte antigen; IA-2 – islet cell antigen 512; ICAM-1 – intercellular adhesion molecule 1; IFNγ – interferon gamma; IFNγR – interferon gamma receptor; IGRP – islet-specific glucose-6-phosphatase catalytic subunit-related protein; IL-1 – interleukin 1; IL-1R – interleukin 1 receptor; KLRG1 – killer cell lectin-like receptor G1; MHC – major histocompability complex; NK cell – natural killer cell; NKT cell – natural killer T cell; NO – nitric oxide; NOD – nonobese diabetic; NOD IL-R – IL-1 receptor-deficient NOD mice; NODTNFR1 – NOD mice deficient in; TNF receptor 1 subunit; nPOD – Network for Pancreatic Organ Donors with Diabetes; PBMC – peripheral blood mononuclear cell; PD-1 – programmed death 1; PDL-1 – programmed death 1 ligand; PLN – pancreatic lymph node; PPI – pre-proinsulin; RIP-LCMV – rat insulin promoter lymphocytic choriomeningitis virus; SOCS1 – suppressor of cytokine signaling 1; T1D – type 1 diabetes; TCR – T cell receptor; Teff – effector T cell; TNFα – tumor necrosis factor alpha; VCAM-1 – vascular cell adhesion molecule 1
1. Introduction
Type 1 diabetes (T1D) is an organ-specific autoimmune disease and represents a failure of self from non-self discrimination, the fundamental task of the immune system. How the immune system goes about this immensely complex task, based both on sequence differences and pattern recognition, is an enduring central question in immunology. While much more is known about the biological mechanisms of the immune system than ever before, our knowledge remains incomplete.
Pathogenic mechanisms are events that are part of the chain of causation of a disease. They can be observed by microscopy or other technologies and follow genetic and environmental etiological factors. In autoimmune diabetes, the expression of perforin in cytotoxic T cells is a decisive pathogenic mechanism because when perforin is deficient diabetes is prevented. However, these mechanisms are not etiological factors as polymorphisms in their genes do not play a critical role. In T1D, pathogenic mechanisms lead to the destruction of pancreatic beta-cells and the eventual presentation of hyperglycemia after months or years of preclinical disease activity. A detailed knowledge of the chain of causative events is critical to our ability to design and implement rational therapies.
An increasingly detailed knowledge of how diabetes progresses is being obtained in parallel with advances in other areas such as therapeutics. A chain of causation implies that causation can be tested, and this requires the use of mouse models. In T1D research, the NOD mouse and all its transgenic variants remain the most commonly used models. Their weakness is that they represent just one genotype, whereas human T1D is much more diverse. There is also the ubiquitous risk of self-congratulation about our understanding of diabetes in mice, while our knowledge of human disease is much more limited.
This review describes the immune-mediated pathogenic mechanisms leading to diabetes. The data described is primarily in the NOD mouse, but also in humans where available. This includes activation, the mechanisms used to induce beta-cell death, and events beyond this point. In particular, we discuss the important roles the islet environment and beta-cell play in propagating the immune response, which ultimately results in beta-cell destruction and disease.
1.1 T lymphocytes destroy beta-cells
Beta-cell destruction in T1D is principally mediated by T lymphocytes. Autoreactive cytotoxic CD8+ T lymphocytes (CTLs) are the main mediators of beta-cell destruction in both the NOD mouse and humans. Effector CD4+ T cells also have an important role. Several CD8+ and CD4+ T cell clones specific for beta-cell antigens have been isolated from mice. These isolated clones are independently capable of transferring disease to immunocompromised hosts [1, 2]. Similarly, transgenic expression of T cell receptor (TCR) from various CD4+ and CD8+ T cell clones results in accelerated diabetes on the NOD background [3, 4]. However, in non-transgenic mice, both CD8+ and CD4+ T cells are essential for the development of spontaneous diabetes as a deficiency of either prevents the development of insulitis and diabetes [4].
Furthermore, both CD8+ and CD4+ T cells are required for efficient adoptive transfer of disease [2]. Non-depleting anti-CD4 and/or anti-CD8 antibodies can also prevent insulitis and diabetes in young NOD mice, and they can reverse disease in recent-onset NOD mice, through alterations in cytokine production and maintenance of immuno regulation [5, 6]. Collectively, these data demonstrate the importance of both autoreactive CD8+ and CD4+ T cells in mediating spontaneous diabetes.
1.2 Function of other immune cells in beta-cell destruction
Other immune cell subsets that contribute to diabetes include B lymphocytes, macrophages and dendritic cells (DCs). B-cell-derived autoantibodies against various islet antigens including (pro)insulin and glutamic acid decarboxylase 65 (GAD65) are the earliest indicators of beta-cell autoimmunity. They are measurable in peripheral blood, and foreshadow the onset of disease. NOD mice, deficient in B cells via the Igµ null mutation, are completely protected from insulitis and diabetes. Depletion of B cells using anti-CD20 also protects NOD mice, or reduces diabetes, depending on the age of administration [7, 8]. In addition, the skewing of the B cell repertoire towards islet antigens, for example through the transgenic expression of insulin-binding immunoglobulin heavy chains in B cells, promotes the development of diabetes [9].
Despite strong evidence for the requirement of B cells in diabetes, their exact function remains unclear. Secretion of antibodies with direct effects on beta-cells, or the presentation of antigens to CD4+ T cells via MHC class II, are the two proposed B cell functions. Recent studies have clarified this and demonstrated that diabetes cannot be transferred by antibody or B cells alone [10]. Instead, B cells contribute primarily as antigen-presenting cells [10-12]. Using a series of transgenic and mutant mice with increased B cell activation, Silva et al. demonstrated that B cells act to enhance the accumulation and function of islet-reactive CD4+ T cells promoting diabetes development [10]. Macrophages and natural killer (NK) cells have also been proposed as effector cells, recruited to the islet by CD4+ T cells to induce beta-cell death [13, 14]. The primary function of DCs in spontaneous diabetes is antigen presentation both in the periphery and within the islet. This is critical to the initiation and progression of disease [15-18].
2. The pancreatic lymph node
2.1 Autoreactive T cells are primed in the pancreatic lymph nodes
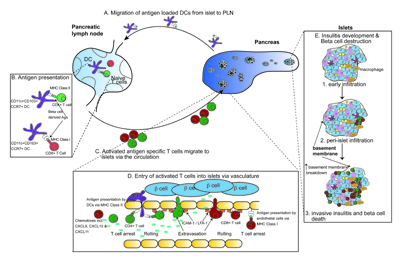
The pancreatic lymph node (PLN) is the site where naïve T cells are activated by islet antigens (Figure 1). T cells that respond to islet antigens can be detected in the PLNs before the onset of insulitis [19]. Adoptively transferred TCR transgenic T cells specific for islet antigens are activated in the pancreatic lymph nodes prior to entering the circulation and infiltrating the islets [15, 19-21]. Activation of adoptively transferred T cells in the PLN are detected from about three weeks of age, suggesting that islet-derived antigens are not presented there until this time [20, 22]. Consistent with this finding, removal of the PLN at 3 weeks of age results in a dramatic decrease in the incidence of diabetes [23]. The initiation of beta-cell-specific T cell responses in the PLN infers that islet antigens are delivered there via the lymphatic circulation. Paradoxically, the islets lack lymphatic circulation, suggesting that delivery may require antigen-transit through lymphatics found at the periphery of islets [24].
 |
 |
Figure 1. Activation, migration, entry, and infiltration of immune cells into islets during spontaneous diabetes. A-C: Dendritic cells loaded with beta-cell antigens migrate from islets to pancreatic lymph nodes and present antigen to naïve autoreactive CD4+ and CD8+ T cells. These T cells undergo initial activation and migrate towards the pancreas via the circulation. D: Entry for CD4+ T cells and CD8+ T cells into islets and presentation (CD8+ T cells). Once arrested, T cells roll and extravasate via integrins and adhesion molecules. E: Insulitis development in islets. Initial macrophage infiltration occurs, followed by T cell infiltration, and the formation of peri-islet insulitis. The islet basement membrane breaks down and invasive insultis with concurrent beta-cell destruction occurs. |
|
Migration of antigen-loaded DCs rather than passive drainage probably provides the passage for antigen delivery from the islets to the PLNs (Figure 1) [25]. DCs loaded with beta-cell-derived proteins can be visualized within the PLNs [26]. Two subsets of DCs have been described within islets, the CD11b+ (CD11c+CD11b+CD103-) and the CD103+ (CD11c+CD11bloCD103+) DCs. The CD103+ subset, but not the CD11b+ subset, expresses CCR7, a chemokine receptor associated with DC migration into draining lymph nodes [27, 28]. CD103+ DCs were numerically deficient in the PLNs of CCR7 knockout mice, suggesting that CD103+ DC represents the major migratory population derived from islets (Figure 1) [28]. DCs presenting beta-cell proteins to CD4+ T cells in the PLNs have been described as CD11c+CD11b variable [26]. The DCs that cross-present beta-cell proteins in the PLNs have been described as CD11c+CD8+ [15], CD11c+CD103+CD11b+/- [28], or CD11c+CD11b-/lo merocytic DCs [29]. Merocytic DCs have been associated with induction of T1D [29]. Notably, CD8 expression was not detected on islets or merocytic DCs, but on cross-tolerizing DCs [15]. Given this phenotypic distinction, it is possible that different DC populations are involved in the induction of disease (CD8- DC) and cross-tolerance (CD8+ DC).
The response to islet antigen is affected by the phenotype, maturity, and activation status of the DCs. Normally, presentation of beta-cell antigens in the PLNs can lead to T cell depletion after an initial period of expansion [15, 30]. An imbalance in the DC subsets within the NOD mouse may impair this process of depletion. Defective cross-tolerization in NOD mice has been associated with a genetically determined deficiency in cross-tolerizing CD8+ DC [31, 32]. A key determinant of initiation of a pathogenic CD8 T cell response is the provision of CD4 T cell help and the licensing of DCs [33]. Transgenic NOD mice that express the costimulatory molecule B7-1 (CD80) on beta-cells develop accelerated autoimmunity and beta-cell destruction without a need for CD4+ T cells [34]. This indicates that CD4+ T cells are required in non-transgenic NOD mice (when costimulatory molecules are not expressed by beta-cells) to license DCs to facilitate the activation of CD8+ T cells [34]. This is consistent with CD4+ T cells licensing DCs.
2.2 Insulin is the primary antigen in NOD mice
The autoreactive T cells and antibodies that develop during the course of NOD diabetes target several proteins expressed by beta-cells, including proinsulin, islet cell antigen 512 (IA-2), GAD, islet-specific glucose-6-phosphatase catalytic subunit-related protein (IGRP), zinc transporter 8, and chromagranin A [35-39]. Of these proteins, proinsulin is central to the development of diabetes. However, other studies favor a role for antigens derived from neuroendocrine tissues that surround the islets, for example glial fibrillary acidic protein (GFAP) expressed by peri-islet Swann cells as initiating antigen upstream of insulin [40].
Perturbations of proinsulin expression are uniquely able to alter the course of diabetes in NOD mice. Transgenic overexpression of Ins2, with the aim of inducing proinsulin-specific tolerance, prevents insulitis and T1D [38, 41]. Concomitant induction of tolerance IGRP-reactive T cells of these mice provided direct evidence that the response against IGRP is downstream of that to proinsulin [36]. Conversely, despite the respective induction of tolerance in IGRP- and GAD-specific T cells by overexpression of IGRP or GAD, diabetes was not prevented [36, 37].
Gene knockout experiments similarly indicated a central role for insulin in diabetes. Insulin is encoded by two genes in mice: Ins1 and Ins2, with the latter being the dominant form expressed in the thymus. Knockouts of the Ins1 or Ins2 genes respectively retard or accelerate diabetes [42, 43]. Knockout of both Ins1 and Ins2, with restoration of hormonal function by transgenic expression of insulin, with an inactivated CD4 and CD8 epitope, completely prevents the development of anti-insulin antibodies, insulitis, and diabetes [44]. Knockouts of GAD65 or IA-2 expression does not alter diabetes onset in NOD mice [45, 46]. These findings are consistent with the hypothesis that proinsulin is the primary antigen that initiates disease.
It is clear that CD4+ T cell responses against the insulin B9-23 epitope are essential for diabetes to develop in NOD mice. Evidence for this observation comes from Unanue's lab. They identified two types of CD4+ T cells recognizing insulin, "type A" T cells that respond to the intact insulin protein and the B9-23 peptide and "type B" T cells that only respond to the B9-23 peptide [17, 47]. Importantly, they showed that only type B T cells are pathogenic in NOD mice [48]. This surprising finding is largely explained by the fact that the insulin B9-23 peptide can bind to I-Ag7 in at least two overlapping registers [35, 48]. The minimal epitope B13-21 binds to I-Ag7 with high avidity. It is the predominant register presented when the epitope is derived from intact protein, and it is recognized by type A T cells. The pathogenic type B T cells recognize I-Ag7 with the B9-23-derived peptide bound in a register that is shifted by one amino acid, presenting the minimal epitope of B12-20. This peptide-MHC complex is unstable and only occurs in later endosomes, in the presence of free peptide [48]. Based on this finding, Unanue and colleagues suggest that pathogenic T cell responses against insulin arise in the NOD mouse because type A-specific CD4+ T cells, but not type B-specific CD4+ T cells, are deleted in the thymus where the insulin B13-21/I-Ag7 complexes are abundant [48]. However, in pancreatic islets, where the local concentration of insulin and free insulin B chain peptides is relatively high, sufficient insulin B12-20 complexes form to activate type B insulin B9-23-specific CD4+ T cells, which escape thymic deletion [48].
2.3 Antigen specificity of autoreactive T cells in human type 1 diabetes
Current evidence suggests that T cell responses to proinsulin are also essential for the development of T1D in humans [49, 50]. The T1D susceptibility locus, IDDM2, maps to a variable number of tandem repeats upstream of the proinsulin gene and is believed to regulate its expression in the thymus, which in turn affects central tolerance to proinsulin [51, 52]. Peripheral blood mononuclear cells (PBMCs) from T1D patients produce interferon γ (IFNγ) when stimulated with proinsulin peptides, whereas PBMC from HLA-matched healthy subjects produce IL-10.
CD8+ T cell responses to proinsulin peptides have been measured by ELIspot in PBMCs of patients with T1D, but not HLA-matched healthy control subjects [53]. Human CD8+ T cell clones specific for an epitope in the leader sequence of human pre-proinsulin (PPI) have been isolated. A PPI-derived peptide presented by HLA-A2 (15-23) was identified by elution and mass spectrometry. Skowera et al. were then able to isolate CD8+ T cell clones specific for PPI15-23 and to identify that responses to this epitope were more readily detected in people with T1D than in HLA-matched healthy donors [54]. Recent X-ray crystallography revealed the interaction between HLA-A2/PPI15-23 peptide/TCR [55]. Most attempts to define CD8+ T cell responses have focused on HLA A2, but recently an HLA A24-restricted epitope derived from proinsulin, PPI3-11, has been identified using a similar peptide elution approach [56].
The specificity of human autoimmune CD4+ T cell responses against proinsulin is described in detail. The development of efficient CFSE-based cloning methods facilitates the isolation of human CD4+ T cell clones specific for proinsulin from the peripheral blood of individuals with established T1D [57]. Interestingly, there is a second overlapping epitope, presumably presented in a different register [58]. T cell responses to several other islet antigens are also associated with immune-mediated destruction of the insulin-producing beta-cells. These mechanisms have been extensively reviewed elsewhere [59].
The possibility that T cell epitopes formed by posttranslational modification are the targets of pathogenic T-cell responses adds a new dimension to the analysis of autoimmune T cell specificity. At this point, no single epitope formed by posttranslational modification has been shown to drive beta-cell autoimmunity. The pathogenic NOD mouse T cell receptor, BDC2.5, has been shown to recognize chromogranin A [35]. More recent data suggests that the epitope recognized by BDC2.5 is formed by transglutaminase-mediated glutamine deamidation [60]. In humans, CD4+ T cell responses against an epitope derived from the A chain of human insulin depend on posttranslational modification. In this case, the modification is a vicinal disulfide bond between adjacent cysteine residues. This modification occurs spontaneously during the processing of intact proinsulin [57].
3. Migration of effector cells from lymph nodes to islets
3.1 Islet-infiltrating T cells are antigen-specific
Once activated in PLNs, T cells migrate via the circulation to the islets (Figure 1). Only activated, but not naïve, beta-cell-specific T cells are able to infiltrate non-inflamed islets. This is consistent with a requirement for T cell activation in PLNs for insulitis to occur [61-63]. Inflamed islets may allow the entry of some non-specific and non-activated T cells, but ultimately the T cells that accumulate within the islet lesion are predominantly islet-antigen-specific [62, 64, 65]. The selection of antigen-specific T cells is likely favored both by cognate antigen enhancement of binding and entry from the blood vessels, as well as cognate antigen-dependent expansion within the islets (see below).
3.2 Entry of immune cells into the islets
The initial infiltration of the islets requires recognition of antigen in the islets, thereby favoring entry of islet-specific T cells [61-63]. Microscopic analysis suggests that such cognate interactions may occur at the blood-islet interface [61-63]. In vitro analysis showed that insulin-specific CD8 T cells kill islet-endothelial cells in a cognate manner, suggesting that islet endothelial cells may cross-present insulin to CD8 T cells enabling binding and integrin activation [63]. Whilst endothelial cells are deficient in class II molecules, islet DCs (class II+) are intimately associated with the blood vessels. They frequently extend dendrites into the vessel lumen and provide a point of contact for CD4 T cells [28, 61]. The efficacy of CD4 T cell localization is further enhanced by interactions with intercellular adhesion molecule 1 (ICAM-1) (Figure 1) [62].
Once infiltration has commenced, the islet becomes more receptive to further infiltration and progression of disease [61, 62, 66, 67]. The role of integrins, selectins, and chemokines in this process has been comprehensively reviewed [68]. Both infiltrating immune cells and resident islet cells are involved. The islet itself is an important modifier of outcome. Infiltration by islet-specific CD4 T cells induces the upregulation of ICAM-1 and vascular cell adhesion molecule 1 (VCAM-1) in blood vessels, and ICAM-1 on beta-cells and DCs [61, 62]. Furthermore, insulitis triggers IFN-response genes. Many of these genes are expressed by the non-leukocyte component of the islet [61, 62, 66]. Chemokines are prominent among the induced genes, including Cxcl9, Cxcl10, and Cxcl11 which attract T cells via the CXCR3 receptor [61, 62, 66].
4. Insulitis
4.1 NOD mouse insulitis
As diabetes progresses, the islets become infiltrated by immune cells. This process is termed insulitis (Figure 1). Histological analysis has shown that macrophages and DCs are dominant in the early spontaneous infiltration of NOD islets [69, 70]. The detection of cells that express CD11c or F4/80 in T cell-deficient NOD.scid islets has been used to assert that DCs and macrophages infiltrate islets first in the absence of T cells [69]. However, these cells are sparse, and it is now recognized that DCs resident within healthy islets express these same markers [16, 28]. Increasing islet infiltration and other changes associated with inflammation result in an increase in DC number, suggesting additional DC infiltration during disease progression [16]. Though rare, some T cells are detected in early NOD islet infiltrates, suggesting that these could be the first cells to infiltrate the islets [69, 71].
Initial insulitis is considered benign and is visualized around the periphery of individual islets. It seems probable that this peri-islet infiltrate originates from the islet-efferent blood vessels that form a network at the periphery of the islets, consisting of efferent capillaries converging at post-capillary venules [72]. The progression from benign peri-islet infiltrate to islet invasion and beta-cell destruction is poorly understood. However, it is widely believed that the destructive infiltrate originates from the peri-islet infiltrate. It has been proposed that the peri-islet basement membrane acts as a physical barrier to leukocyte migration into the islets. Histological examination in NOD mice showed that progression to destructive insulitis is associated with destruction of the peri-islet basement membrane (Figure 1) [73].
Recently, the islets of NOD mice have been shown to express high levels of the glycosaminoglycan, heparan sulfate, which is essential for beta-cell survival. Immune cells infiltrating the islets were also shown to produce heparanase, an active herparan sulfate-degrading enzyme, which could act to promote invasive insulitis [74]. Similarly, the local upregulation of hyaluronic acid could also drive the progression to destructive insulitis by its interaction with CD44 on diabetogenic cells [67]. This assertion was supported by the blockade of adoptive transfer of diabetes by in vivo administration of either anti-CD44 mAb or hyaluronidase [67]. Alternatively, following adoptive transfer of activated beta-cell-specific CD4 T cells, it was concluded that these T cells enter the islets directly from the circulation [61]. Whatever mechanisms are involved, it is clear that insulitis in the NOD mouse follows a distinct and well characterized pattern; it presents as an early peri-islet insulitis that progressively develops to destructive insulitis with concurrent beta-cell destruction.
4.2 Insulitis in human islets
Insulitis is also a feature of the development of diabetes in humans. Histopathological analysis of pancreata collected at or near diagnosis revealed infiltration of T cells, B cells, and macrophages into the islets [75]. CD8+ T cells are the most abundant leukocytes, while CD4+ T cells are less in number than both CD8+ T cells and macrophages [76-78]. The infiltration of islets is often accompanied by a marked upregulation of MHC class I protein on islets under immune attack [76, 78]. This and the prevalence of CD8+ T cells within the islet lesions, suggests that CD8+ T cells are the dominant effector cells in human diabetes.
Insulitis in humans differs in several features from that of the NOD mouse. One of the most striking differences is that human islet infiltration and destruction is not uniform or synchronous. Frequently, it only affects a small number of islets, and lobular areas of the pancreas are affected differently [77]. A recent study of pancreata collected from autoantibody positive pre-clinical patients demonstrated the presence of insulitis in 2 out of 62 patients, and <10% of the islets studied within these two samples were affected [79]. Similarly, a study of recent onset diabetic pancreata detected small amounts of infiltrate, with a few immune cells per islet [80]. Human insulitis is qualitatively similar to that in the NOD mouse with a similar mix of cell types, but it is clearly not as florid as in the mouse. However, studies of pancreata collected from long standing diabetics have shown that infiltrate can persist for a considerable length of time, suggesting it is an important feature of the human disease [81].
Analysis of insulitis and effector cells within the pancreas is hampered by the difficulties in obtaining samples for study. However, progress on the direct study of effector cells within the pancreas has been made recently. An elegant study by Coppietiers et al. utilized a large collection of frozen pancreas samples collected through the Network for Pancreatic Organ Donors with Diabetes (nPOD), combined with some other rare cases obtained by the authors, to demonstrate definitively the presence of islet-reactive CD8+ T cells within the islets of type 1 diabetic patients [82]. The authors used in situ tetramer staining of sections collected from HLA-A2 individuals with up to 8 years disease duration, to show specific reactivity of CD8+ T cells within the insulitic lesion to six islet autoantigens, PPI, insulin, IGRP, GAD65, preproislet amyloid protein, and IA-2 [82]. These studies are important as they analyze directly the antigen specificity of T cells at the site of autoimmune beta-cell destruction. Within the limitations of human research, this provides excellent evidence for these T cell responses playing a direct role in beta-cell destruction.
5. Mechanisms of beta-cell destruction
5.1 The requirement for CD8+ T cells
Autoreactive cytotoxic CD8+ CTLs are dominant in destroying beta-cells. CTLs recognize peptide antigens presented on the beta-cell surface by MHC class I proteins that consist of a polymorphic heavy chain and a constant light chain, β2microglobulin. NOD mice deficient in β2-microglobulin lack MHC class I and CD8+ T cells and are protected from diabetes [83-86]. Reconstitution of the beta-cell MHC class I expression results in diabetes following the transfer of diabetogenic splenocytes [87].
These experiments indicate a need for the direct interaction between beta-cell and CD8+ T cell via MHC class I for beta-cell destruction. Overexpression of adenovirus E19 protein in beta-cells, which inhibits MHC class I expression, prevents CTL lysis [19]. Transgenic overexpression of the suppressor of cytokine signaling 1 (SOCS1) in beta-cells blocks CTL killing of beta-cells in part by reducing class I MHC expression and antigen presentation [88, 89]. However, the generation of NOD mice, containing a conditional deletion of class I specifically from the beta-cells (class I beta-bald mice), directly confirm the interaction between CTL and beta-cell is required for efficient disease progression [90].
Normally, CD8+ T cells develop in these mice as MHC class I is expressed in the thymus and lymph nodes, but diabetes is significantly reduced. The direct interaction of CD8+ T cell and beta-cell has recently been visualized using intra-vital 2-photon imaging [91]. Tracking of CTL movement into and within the pancreas in the RIP-LCMV model of diabetes, showed CTLs arresting at the junction with beta-cells and forming stable contacts of several hours duration [91]. Collectively, these studies demonstrate the dominant role of CTLs in beta-cell destruction. However, the lack of diabetes prevention in the class I beta bald mice indicates other type of immune cells, including CD4+ T cells can also function as effectors [90].
5.2 Perforin and granzymes released by CTLs are the dominant effectors of beta-cell destruction
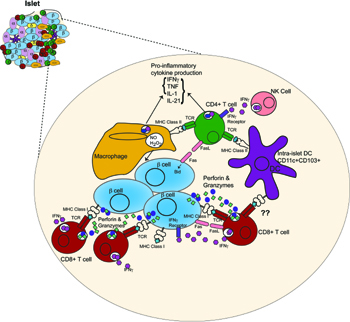
Granule exocytosis with release of perforin and granzymes is a major effector mechanism utilized by all CTLs. Diabetes incidence in NOD mice deficient in perforin was reduced to 17% compared to wild-type littermates and onset was delayed [92]. This observation was independently confirmed [93]. In both studies, a small percentage of mice still developed diabetes, which could be caused by other effector mechanisms or other effector cells. 8.3 TCR transgenic CTLs were reported to kill beta-cells independently of perforin, using the Fas/FasL pathway, suggesting perforin is not the dominant effector mechanism [93]. This was later clarified by Dudek et al. who used genetic deficiencies in both pathways to demonstrate in the 8.3 model that both perforin and Fas are operational and compensate for each other when one effector mechanism is removed [89]. However there is little evidence for this redundancy in NOD mice (see below). Overall the significant effect of perforin deficiency points overwhelmingly to its dominant role in CTL-mediated beta-cell destruction in vivo (Figure 2, Table 1). Perforin deficiency does not reduce diabetes mediated by CD4+ T cells, demonstrating this is unlikely to be a dominant effector mechanism utilized by CD4+ T cells [94, 95].
 |
|
Figure 2. Effector cells and their actions in mediating beta-cell destruction. Cytotoxic T lymphocytes are the main mediators of beta-cell death through the production of perforin and granzymes. Fas-FasL interactions have a minor role in beta-cell destruction. CD4+ T cells do not directly kill beta-cells, instead they are proposed to activate macrophages to kill beta-cell cells via NO and reactive oxygen species. Pro-inflammatory cytokines are produced by CD4+, CTLs, and macrophages, and act to regulate the immune response. The presence of NK cells within the islets is proposed to promote effector CD4+ T cell function. Intra-islet DCs present antigen to CD4+ T cells and may present antigen to CTLs. B cells are also present in the islet and secrete autoantibodies and promote CD4+ T cell function through antigen presentation (not shown). Figure is prepared from descriptions within mouse models. |
|
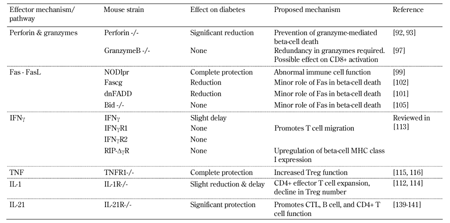
Table
1.
Effects of genetic mutations within effector pathways on diabetes in NOD mice |
|
 |
|
Legend:
Bid – BH3-interacting domain death agonist, CTL – cytotoxic T lymphocyte, dnFADD – dominant-negative Fas-associated death domain, Fascg – dominant-negative form of Fas. IFNγ – interferon gamma, IFNγR1/2 – interferon gamma receptor 1/2, MHC – major histocompatibility complex, NOD – nonobese diabetic, NODlpr – CD95-deficient variant (lpr) of the NOD mouse, RIP – rat insulin promoter, TNF – tumor necrosis factor, TNFR1 – tumor necrosis factor receptor 1. |
|
The main function of perforin is to facilitate the activation and delivery of granzymes into the target cell. Granzymes are a family of serine proteases that activate pathways within the cell to induce apoptosis. Recombinant granzymeB was able to induce beta-cell death in vitro by a BH3-interacting domain death agonist (Bid) dependent mechanism [96]. However, granzymeB was only effective in the presence of perforin. Given the dependence on perforin for granzyme activity, the perforin knockout mouse is also an effective knockout of granzymes. To investigate the role of granzymeB specifically, granzymeB knockout NOD mice were recently generated (Table 1) [97]. Spontaneous diabetes in these mice was unaffected, although insulitis onset was delayed, indicating granzymeB is dispensable for diabetes, but may play a role in CD8+ T cell activation. This suggests a redundancy for granzyme-mediated beta-cell killing, which is perhaps not surprising given that 11 different granzymes are present in mice and expression varies in CTL.
5.3 The Fas/FasL pathway is a minor effector of beta-cell destruction
Ligation of the Fas (CD95) death receptor by FasL on both effector CTLs and CD4+ T cells is another proposed mechanism of beta-cell destruction (Figure 2). Beta-cells have low levels of Fas, but this can increase with exposure to inflammatory cytokines [98]. Ligation of Fas by FasL induces caspase activation and beta-cell apoptosis. FasL-induced beta-cell destruction has been characterized best in vitro. However, its pathogenic role in diabetes has been challenged by several studies. Fas-deficient NOD mice (NODlpr) were resistant to diabetes [99]. However, Fas-deficient islets transplanted into wild-type mice revealed only marginal protection of the islet graft, indicating a minor role for Fas in beta-cell destruction [100]. Transgenic NOD mice, expressing dominant negative Fas-associated death domain (FADD), had limited protection (Table 1). FADD is a component of the Fas signaling pathway that only occurs in beta-cells and transgenic mice containing a beta-cell-specific disruption of Fas [101, 102]. Similarly, islets deficient in Fas expression were not protected in CD4 T cell-induced diabetes, suggesting beta-cell destruction occurs in the absence of a functional Fas pathway [103, 104].
A drawback of these models is the overexpresssion of transgenes in beta-cells which can affect beta-cell function. To circumvent this, Mollah et al. investigated the incidence of diabetes in NOD mice deficient in Bid [105]. Bid is essential for Fas-induced apoptosis and Bid-deficient islets are protected from FasL-induced killing in vitro [106]. Development of spontaneous diabetes in NOD Bid-/- mice was similar to wild-type mice, indicating Bid-dependent Fas-induced apoptosis is not an effector mechanism utilized by T cells in the NOD mouse. Collectively, the impact of any of the mutations in the Fas-FasL pathway is relatively minor, demonstrating this pathway has only a small role in beta-cell destruction mediated by either CTLs or CD4+ T cells (Table 1).
5.4 Effector mechanisms utilized by human auto-reactive CTLs
CTL clones recognizing several islet autoantigens have been isolated from the peripheral blood of both recent onset and long-term T1D patients [54, 107]. The increased expression of beta-cell MHC class I observed in pancreatic biopsies suggests that a direct interaction between CTLs and beta-cells is required for CTL killing in humans [75, 76, 82, 108]. Skowera et al. demonstrated that their isolated CTL clone specific for PPI15-23 was able to kill beta-cells and required direct contact between CTL and beta-cell. This indicates that perforin or Fas are the likely candidate effector molecules.
Human islets are susceptible to killing by recombinant perforin and granzymeB in vitro [96]. To investigate which CTL effector mechanisms are utilized to kill beta-cells Campbell et al. used CTL clones specific for viral peptides presented by HLA-A2, HLA-B8, and human beta-cells pulsed with specific viral peptides [109]. This system circumvents the difficulties in isolating beta-cell antigen-specific CTLs. The CTLs killed beta-cells pulsed with viral peptides, dependent on MHC class I expression. Increasing the expression of MHC class I on the beta-cell surface increased the CTL killing. Beta-cell death was prevented by the presence of the perforin inhibitor concanamycin A. Therefore, killing of beta-cells was dependent on perforin. In contrast, neutralizing anti-Fas ligand antibody had no effect, suggesting the Fas-FasL pathway is not involved [108].
These findings have recently been confirmed using the PPI15-24 clone and an additional HLA-A2-restricted PPI clone, PPI3-11 [106]. These clones killed beta-cells by requiring a direct interaction via MHC class I and utilizing perforin and granzymes. The role of Fas/FasL was less clear, and there was no evidence for cytokine-dependent killing [106]. Overall, these studies demonstrate autoreactive CTLs in humans utilize the same effector mechanisms as those in the NOD mouse. There is a dominant role for perforin and granzymes, and a possible minor role for Fas in mediating beta-cell death.
5.5 Pro-inflammatory cytokines are critical regulators of effector function, but they are not important effectors of beta-cell destruction
Whilst CD8+ T cells are the dominant effector cells, CD4+ T cells are also involved in beta-cell destruction (Figure 2), even though beta-cells do not express MHC class II, and thus do not interact with CD4 T cells directly. Several mechanisms have been proposed to explain how CD4+ T cells destroy beta-cells. Studies have suggested inflammatory cytokines such as IFNγ, TNFα, and IL-1 are important mediators of beta-cell death induced by CD4+ T cells [110]. Pro-inflammatory cytokines are also secreted by activated CTLs. Treatment of islets with IFNγ and IL-1, or IFNγ and TNFα, leads to beta-cell death in vitro, probably through production of nitric oxide (NO) in beta-cells and activation of the intrinsic death machinery regulated by Bcl-2 family members [110, 111]. However, the in vivo role of inflammatory cytokines in beta-cell death remains controversial; extensive studies indicate that cytokines are not directly cytotoxic to beta-cells [103, 112, 113]. Instead, the actions of these cytokines are to mediate intracellular communication, recruit immune cells to islets, and/or increase beta-cell recognition by autoreactive CTLs (Table 1).
Experiments in knockout mice have shown that deficiencies in single cytokines or their receptors usually do not prevent diabetes. When they do, it is due to immunoregulatory effects (Table 1). Mice deficient in IFNγ or the IFNγ receptor subunits, IFNγR1 or IFNγR2, develop diabetes, demonstrating IFNγ is not required for spontaneous diabetes in NOD mice (reviewed in [113]). The onset of diabetes is also unaffected in NOD mice expressing a dominant negative form of IFNγR1 in the beta-cells (RIP-ΔγR NOD mice) (reviewed in [113]). IFNγ is responsible for the upregulation of MHC class I on beta-cells. This is observed in islets with insulitis in NOD mice, which promotes extravasation of CD4+ and CD8+ T cells through the vessel endothelium into islets (reviewed in [103, 113]). Similarly, the incidence of diabetes in IL-1 receptor-deficient NOD mice (NOD IL-R) is only slightly reduced and delayed. Comparing the destruction of wild-type and IL-1R-deficient islets in transplant experiments revealed that the IL-1R deficiency impacts on immune cells rather than beta-cells [112]. NOD mice express elevated levels of IL-1, which promotes the expansion of effector CD4+ T cells and dilutes the number of CD4+CD25+ regulatory T cells (Tregs) [114]. In contrast, NOD mice deficient in the TNF receptor 1 subunit (NODTNFR1) are completely protected from diabetes, and develop a very mild peri-islet insulitis [115]. TNFR1 deficiency affects the immune system rather than protecting islets directly [116, 117]. Tregs from TNFR1-deficient mice are better able to suppress effector cells both in vitro and in vivo [116].
Blockade of several cytokines by overexpression of SOCS1 in beta-cells has a greater impact on diabetes mediated by CD8+ T cells than a single cytokine deficiency has on diabetes development [88]. SOCS1 blocks signaling by multiple cytokines, including IFNγ. This approach completely prevents diabetes in the 8.3 CD8+ T cell model, an effect linked to a reduction in beta-cell recognition by CD8+ T cells [88, 89]. This result demonstrates one way in which the beta-cell contributes to its own demise. However, SOCS1 overexpression does not protect against CD4+ T cell-induced diabetes [103]. Furthermore, IL-1 receptor knockout beta-cells overexpressing SOCS1 are destroyed in vivo as fast as wild-type beta-cells in mice carrying diabetogenic CD4+ T cells [103]. In fact, only systemic, but not beta-cell-specific, deletion of cytokine signaling can delay or inhibit the development of CD4+ or CD8+ T cell-induced diabetes. This shows that inflammatory cytokines are critical regulators of the immune response, but are not required for beta-cell death.
5.6 The function of human autoreactive CD4+ T cells
The strongest evidence for a role of CD4+ T cells in humans originates from the genetic association between HLA class II region genes and T1D. Genetically, the HLA class II region has the strongest association with T1D. More than 90% of Caucasians with T1D carry the DR3-DQ2 (HLA DRB1*0301-DQB1*0201) or DR4-DQ8 (HLA DRB1*0401-DQB1*0302) haplotypes [118]. Surprisingly, individuals heterozygous for DR3-DQ2 and DR4-DQ8 haplotypes are at greater risk of T1D than those with either one of these haplotypes alone [119]. The role of CD4+ T cells in human T1D is underscored by the observation that some HLA alleles, HLA DQB1*0602 for example, confer a reduced risk of T1D [120].
The effector mechanisms used by pathogenic human CD4+ T cells are not fully understood, primarily because of the technical challenges associated with identifying beta-cell antigen-specific T cells in the peripheral blood [121]. The issues surrounding the identity of the antigens and epitopes recognized by pathogenic CD4+ T cells add to the challenge. Nonetheless, it has been shown that the CD4/CD8 ratio is altered in patients with type 1 diabetes, and that CD4+ T cells in patients means an increase in their activation status [122, 123].
Further studies with human T cells have shown that CD4+ T cells with a Th17 phenotype are increased in T1D patients, with a concomitant increase in the concentration of IL-17 detectable in peripheral blood [124-127]. In addition, Treg function is decreased in T1D patients. Brusko et al. detected no change in the number of Tregs [128]. However, the suppressive function of Tregs isolated from patients is impaired, which may contribute to the loss of self-tolerance [126, 128, 129]. Collectively, declining Treg function and increasing effector CD4+ T cell number could promote diabetes pathogenesis.
5.7 The emerging role of macrophages and NK cells
Evidence exists that macrophages take a decisive role in the destruction of pancreatic beta-cells. CD4+ T cells may activate islet-infiltrating macrophages that then kill beta-cells (Figure 2). Diabetogenic CD4+ T cells fail to induce diabetes in the absence of macrophages. Macrophages are in close proximity to beta-cells in insulitis. Depletion of macrophages using clodronate-loaded liposomes inhibits diabetes induced by diabetogenic CD4+ T cells [13]. Activated macrophages isolated from infiltrated islets kill islet cells in vitro and in vivo [13]. Furthermore, the accumulation of macrophages within islets following chemokine expression by beta-cells can cause beta-cell death and diabetes, even in the absence of T cells [130]. Macrophages can directly exert cytotoxic effects through a variety of mechanisms. They are an important source of inflammatory cytokines, NO, and reactive oxygen species such as H2O2, all of which kill beta-cells in vitro [110]. Macrophages also have the ability to sense stressed cells and eliminate them by engulfment [131]. However, the in vivo mechanism of macrophage-mediated killing of beta-cells has not been established and remains an intriguing question.
Natural killer cells seem to play another complementary role in beta-cell destruction in autoimmune diabetes (Figure 2). It has been reported that a ligand for the NK cell receptor, NKp46, is expressed by beta-cells [14]. Interaction of NK cells and beta-cells could lead to the release of cytotoxic molecules from NK cells such as perforin and lysis of beta-cells. However, Angstetra et al. were unable to demonstrate that NK cells were directly cytotoxic to beta-cells, either in vitro or in vivo [132]. Instead, the study showed that depletion of NK cells in NOD4.1 mice reduced the incidence of diabetes, suggesting NK cells could maintain the right environment for the cytotoxicity of effector T cells [133]. This is consistent with an earlier report showing that the ablation of Tregs within islets results in activation of NK cells and the production of IFNγ, which promotes the function of effector CD4+ T cells [133]. These two studies clearly indicate that the function of NK cells is to promote effector T cell function, as opposed to being directly cytotoxic to beta-cells (Figure 2).
6. The function of the islet environment
Recent studies have revealed that the progression of diabetes is driven by pathogenic events that occur within the islet microenvironment, including differentiation and proliferation of T cells. This suggests that the islet is not simply a passive target of autoimmune effector cells that develop within the lymph node.
6.1 Immune regulatory mechanisms within islets fail to control progression of insulitis
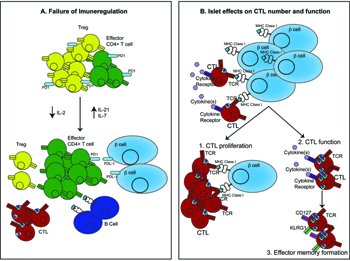
Tregs are readily identified within the sites of inflammation. The maintenance of Tregs at the site of inflammation is not simply the result of continued migration from lymphoid tissues. The inflammatory lesion also provides signals for Treg proliferation and survival, and Ki67+FOXP3+ Tregs have been detected within islets [26]. However, the progression of insulitis has been associated with a progressive reduction in the ratio of Tregs to effector T cells (Teffs) within islets [134]. While the events that lead to the failure of intra-islet Tregs to control insulitis remain to be fully elucidated, one contributor may be the insufficient local availability of IL-2 resulting in decreased Treg expression of Bcl2 and consequent apoptosis [134]. The NOD Il2 gene is associated with reduced IL-2 production and reduced Treg number and function compared to the Il2 gene of T1D resistant strains [135]. In addition, CD4+ Teffs may acquire resistance to regulation, which further contributes to disease progression (Figure 3) [136]. Altered responsiveness of Tregs to IL-2 has also been observed in patients with T1D, and this leads to a decrease in the persistence of FoxP3 expression, declining function, and autoimmunity [137].
 |
|
Figure 3. How the islet environment modifies the pathogenesis of diabetes. A: Immune regulatory mechanisms fail within the islet as diabetes progresses. Tregs present in the islet initially control the immune response. However, declining concentrations of IL-2 and increasing concentration of IL-21 result in decreased Treg number and function and increases in effector CD4+ T cells. The high concentration of IL-21 also promotes CTL and B cell function in the islet. Increasing concentrations of IL-7 results in downregulation of PD-1 expression on Tregs and effector CD4+ T cells, which promotes beta-cell destruction through failure of the PD-1/PDL-1 pathway. B: Islet environment promotes CTL function. Beta-cell antigen presentation promotes local proliferation of CTLs within the islet, increasing insulitis development. CTLs also respond to cytokines present in the islet, and this may increase the expression of cytotoxic markers, including granzymeB and IFNγ, and CTL function. CTLs within the islet are also stimulated to form an effector memory CTL pool. |
|
Reduced local IL-2 production is linked with high levels of IL-21 secretion within the islets of NOD mice. The Idd3 locus contains both the Il2 and Il21 genes, and both are highly expressed in NOD mice. However, the IL-2 mRNA is unstable, resulting in low level expression and a high level of IL-21 [138]. IL-21 is highly expressed by activated CD4+ T cells and NKT cells. Deficiency in IL-21 signaling, either through genetic deficiency of the IL-21 receptor or through IL-21 neutralization prevents diabetes and insulitis [139-141]. Neutralization of IL-21 alters the function of CD8+ T cells and B cells within the islet, suggesting this cytokine promotes CTL and B cell differentiation, and survival within islets [139]. Expression of IL-21 may also stimulate expression of CCR7 on DCs. This facilitates the migration of antigen-loaded DCs from islets into the PLN, where they can cross-present antigens to autoreactive CD8+ T cells [27]. Thus, the imbalance between IL-2 and IL-21 within the islet environment results in declining Treg function, with a concomitant increase in effector T cell function (Figure 3).
Another common gamma chain cytokine, IL-7, may promote T cell function and progression of diabetes. Anti-IL-7 antibody treatment prevented the development of spontaneous disease in NOD mice, and even reversed established disease [142, 143]. IL-7 neutralization appeared to increase the proportion of effector cells and Tregs expressing the inhibitory cell surface receptor programmed death 1 (PD-1) [142, 143]. PD-1 binds its ligand PDL-1 expressed on lymphocytes and beta-cells. The interaction between PD-1 and PDL-1 has previously been shown to be very effective at preventing diabetes [144, 145]. Therefore, the expression of IL-7 within the islets may suppress PD-1/PDL-1 interaction and promote pathogenic T cells to kill beta-cells (Figure 3).
6.2 Antigen presentation within the islet environment
There are at least four cell types within infiltrated islets that could act to present beta-cell antigens to CD4+ and CD8+ T cells. These include dendritic cells, endothelial cells, B cells, and the beta-cells themselves It is likely that the presentation to either CD4+ or CD8+ T cells utilizes different cell types. The importance of antigen presentation by beta-cells in CTL destruction is described above (section 5.1), and its role in promoting effector function is described below (sections 6.3 and 6.4). Likewise the function of endothelial cell antigen presentation is described above (section 3.2).
It has been suggested that B cells present antigens to both CD4+ and CD8+ T cells, although it is unclear whether this occurs within the islet environment or just within the PLN [10-12]. B cells have also been shown to promote the survival of autoreactive CTLs within islets. Depletion of B cells resulted in increased CTL apoptosis within islets, and indicated that B cells are required for intra-islet CTL survival and development of diabetes [146]. As described in sections 2.1 and 3.1 above, all islets contain intra islet dendritic cells, and these increase as insulitis progresses. Both CD11b+ (CD11c+CD11b+CD103-) and CD103+ (CD11c+CD11bloCD103+) DCs are present in non-inflammed islets, they are more plentiful in the islets of NOD mice, and the CD11b+ population expands during insulitis [16, 28]. These intra-islet dendritic cells contain beta-cell antigens, including insulin. The ability of these islet DCs to take up and present beta-cell antigens has been demonstrated by the capacity of freshly explanted islet DC to stimulate CD4 T cells [16, 17]). Cross-presentation by intra-islet DCs to CTLs within islets has not been demonstrated.
6.3 The local T cell response is driven by ongoing recruitment and intra-islet proliferation
Progression of insulitis is due both to CD4+ and CD8+ T cell recruitment from the peripheral circulation and proliferation of previously recruited cells within the islet environment. The sphingosine-1-phosphate receptor agonist, FTY720, that prevents the egress of lymphocytes from lymph nodes into the circulation, has been useful in determining which of these processes contributes to insulitis progression. FTY720 treatment effectively disconnects the islets from circulating lymphocytes. Continuous treatment of NOD mice with FTY720 prevents the development of diabetes [147]. Protection in the treated mice is not due to clearance of the islet infiltrate, but rather a stabilization of the existing insulitis. A two to three fold reduction in CD4+ T cells within the islets was observed, but no change was seen in the numbers of infiltrating CD8+ T cells present [147].
A similar approach in NOD8.3 mice identified that for CD8+ T cells both recruitment from the circulation and local proliferation within the islet is required for the progression of insulitis [148]. Antigen presentation by the beta-cell was critical in stimulating CTL proliferation within the islet (Figure 3). These two studies suggest that CD4+ and CD8+ T cells use continuous recruitment and local proliferation differently during insulitis development. This clarifies the importance of the islet environment in enhancing the CD8+ T cell response.
6.4 CTLs are armed within islets
The islet environment also promotes the differentiation of CTLs into effector memory T cells (Figure 3) [149]. Autoreactive CTLs found within the islets of diabetes-prone NOD mice display much higher expression of markers of cytotoxicity than recently activated cells, which are still residing in the PLNs. These markers included granzyme B, IFNγ, and surface expression of the degranulation marker CD107a. Their expression only increased when the CD8+ T cells reached the islets. Pro-inflammatory cytokines appeared to provide the additional signals for CTL maturation rather than antigen presentation by beta-cells or intra-islet dendritic cells [149]. Adoptively transferred 8.3 CD8+ T cells increased their expression of CD127 and KLRG1, two markers of effector memory differentiation. This phenotype was acquired within the islets (Chee, Kay & Krishnamurthy, unpublished 2012). These two studies highlight the importance of the islet in CTL differentiation.
6.5 Post beta-cell destruction events
Our understanding of the events within the islets that occur following beta-cell destruction is limited due to difficulties in studying these events in both the NOD mouse and humans. Maintaining NOD mice post diabetes onset is problematic as they are moribund, and administration of insulin is complicated. In humans, obtaining biopsy specimens from patients is almost impossible. A recent study of diabetic patients whose duration of disease was 50 years or more (Joslin medalists) identified the presence of residual insulin-positive beta-cells [81]. This surprising finding suggests not all beta-cells are destroyed and/or beta-cells can regenerate over time.
Analysis of insulitis reveals that immune infiltration can persist, but it can also resolve leaving glucogon- and somatostatin-postive cells alone in pseudo-islets. Death of the T cells within the islet, once beta-cell destruction had occurred, was the mechanism proposed to cause the resolution of insulitis. However, recent studies have suggested that emigration from islets to the periphery, and formation of the peripheral effector memory T cells pool, is a more likely outcome. T cells specific for GAD65 and preproinsulin displaying an effector memory phenotype have been detected in the peripheral blood of patients [55, 150]. We have recently studied the formation of these cells in the NOD mouse. We found that they are formed within the islet, and then leave the environment to seed the periphery (Chee, Kay & Krishnamurthy, unpublished 2012). This is consistent with the visualization of T cells leaving the islets and with an older observation of autoreactive T cells appearing in the blood in waves [91, 151]. Clearly, we still have much to learn about post beta-cell destruction events in both mice and humans, and this will be the focus of much research in the future.
7. Summary and conclusions
Much has been learned about the sequence of events that leads to type 1 diabetes, so that our current understanding is substantially different to that of a couple of decades ago. Yet, the more that we learn the more we realize we still do not know. In an overview, we understand that a process of aberrant immune recognition of self beta-cells by T cells occurs accompanied by autoantibody production. Immune infiltration of the pancreatic islets proceeds with loss of beta-cells and failure of insulin production. If regeneration of beta-cells occurs, then this is insufficient for metabolic function. Yet, within this schematic outline, we need to know more about the particular details for effective therapy to be developed. It is not possible at this time to clearly describe a current treatment with a very high likelihood of therapeutic benefit. In particular, we need to understand how much the great level of detail known about the NOD mouse is applicable to humans, and to what extent the sequence of pathogenic events varies between individual humans with type 1 diabetes.
Several recent major advances have been made (Table 2). They include identifying the islet as an important location of disease pathogenesis, not merely a passive target of events within the lymph nodes (Figure 3, Table 2). Technological innovation, for example those involving the use of class I and class II MHC tetramers, and advances in microscopy and other forms of imaging are allowing us to revisit and re-conceptualize events. Isolation of effector cell populations from T1D patients, and analysis of pancreatic samples from individuals with type 1 diabetes, mostly after death, are also providing important new information.
The discovery that perforin is the dominant effector mechanism utilized by human autoreactive CTL to kill beta-cells, and the identification of CD8+ T cells specific for known beta-cell antigens present in islets of people with diabetes, are reassuring discoveries, as they mirror mechanisms described in the NOD mouse (Table 2). This overlap shows that analysis of the NOD mouse in conjunction with human samples will continue to be fruitful. We are yet to determine exactly how CD4+ T cells act to kill beta-cells. The NOD model will be essential to understand this and many other questions. The very early pathogenic events remain difficult to study, especially in humans.
Of course, the target of the field is to understand and to treat human type 1 diabetes. Translation of findings from animal models, and a greater focus on studying human diabetes with the latest technology, should yield new insights and new opportunities for an effective future therapy to prevent or cure type 1 diabetes.
Table
2.
Recent major discoveries in the pathogenic mechanisms of type 1 diabetes |
|
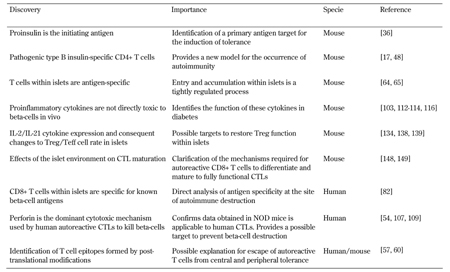 |
|
Legend:
CTL – cytotoxic T lymphocyte, IL-2 – interleukin 2, NOD – nonobese diabetic. |
|
Disclosure: The authors report no conflict of interests.
Acknowledgments:
This work was supported by grants and fellowship from the National Health and Medical Research Council of Australia (NHMRC) and the Juvenile Diabetes Research Foundation (JDRF). St. Vincent's Institute and the Walter and Eliza Hall Institute are supported by the Operational Infrastructure Support Scheme from the Victorian State Government.
References
- Bradley BJ, Haskins K, La Rosa FG, Lafferty KJ. CD8 T cells are not required for islet destruction induced by a CD4+ islet-specific T-cell clone. Diabetes 1992. 41:1603-1608. [DOD] [CrossRef]
- Christianson SW, Shultz LD, Leiter EH. Adoptive transfer of diabetes into immunodeficient NOD-scid/scid mice. Relative contributions of CD4+ and CD8+ T-cells from diabetic versus prediabetic NOD.NON-Thy-1a donors. Diabetes 1993. 42:44-55. [DOD] [CrossRef]
- Verdaguer J, Yoon JW, Anderson B, Averill N, Utsugi T, Park BJ, Santamaria P. Acceleration of spontaneous diabetes in TCR-beta-transgenic nonobese diabetic mice by beta-cell cytotoxic CD8+ T cells expressing identical endogenous TCR-alpha chains. J Immunol 1996. 157:4726-4735. [DOD]
- Wong FS, Janeway CA Jr. The role of CD4 vs. CD8 T cells in IDDM. J Autoimmun 1999. 13:290-295. [DOD] [CrossRef]
- Phillips JM, Parish NM, Raine T, Bland C, Sawyer Y, De La Pena H, Cooke A. Type 1 diabetes development requires both CD4+ and CD8+ T cells and can be reversed by non-depleting antibodies targeting both T cell populations. Rev Diabet Stud 2009. 6(2):97-103. [DOD] [CrossRef]
- Yi Z, Diz R, Martin AJ, Morillon YM, Kline DE, Li L, Wang B, Tisch R. Long-term remission of diabetes in NOD mice is induced by nondepleting anti-CD4 and anti-CD8 antibodies. Diabetes 2012. 61:2871-2880. [DOD] [CrossRef]
- Serreze D. V, Chapman HD, Varnum DS, Hanson MS, Reifsnyder PC, Richard SD, Fleming SA, Leiter EH, Shultz LD. B lymphocytes are essential for the initiation of T cell-mediated autoimmune diabetes: analysis of a new “speed congenic” stock of NOD.Ig mu null mice. J Exp Med 1996. 184:2049-2053. [DOD] [CrossRef]
- Xiu Y, Wong CP, Bouaziz JD, Hamaguchi Y, Wang Y, Pop SM, Tisch RM, Tedder TF. B lymphocyte depletion by CD20 monoclonal antibody prevents diabetes in nonobese diabetic mice despite isotype-specific differences in Fc gamma R effector functions. J Immunol 2008. 180:2863-2875. [DOD]
- Hulbert C, Riseili B, Rojas M, Thomas JW. B cell specificity contributes to the outcome of diabetes in nonobese diabetic mice. J Immunol 2001. 167:5535-5538. [DOD]
- Silva DG, Daley SR, Hogan J, Lee SK, Teh CE, Hu DY, Lam KP, Goodnow CC, Vinuesa CG. Anti-islet autoantibodies trigger autoimmune diabetes in the presence of an increased frequency of islet-reactive CD4 T cells. Diabetes 2011. 60:2102-2111. [DOD] [CrossRef]
- Wong FS, Wen L. B cells in autoimmune diabetes. Rev Diabet Stud 2005. 2(3):121-135. [DOD] [CrossRef]
- Serreze DV, Fleming SA, Chapman HD, Richard SD, Leiter EH, Tisch RM. B lymphocytes are critical antigen-presenting cells for the initiation of T cell-mediated autoimmune diabetes in nonobese diabetic mice. J Immunol 1998. 161:3912-3918. [DOD]
- Calderon B, Suri A, Unanue ER. In CD4+ T-cell-induced diabetes, macrophages are the final effector cells that mediate islet beta-cell killing: studies from an acute model. Am J Pathol 2006. 169:2137-2147. [DOD] [CrossRef]
- Gur C, Porgador A, Elboim M, Gazit R, Mizrahi S, Stern-Ginossar N, Achdout H, Ghadially H, Dor Y, Nir T, et al. The activating receptor NKp46 is essential for the development of type 1 diabetes. Nat Immunol 2010. 11:121-128. [DOD] [CrossRef]
- Belz G. T, Behrens GM, Smith CM, Miller JF, Jones C, Lejon K, Fathman CG, Mueller SN, Shortman K, Carbone FR, Heath WR. The CD8alpha(+) dendritic cell is responsible for inducing peripheral self-tolerance to tissue-associated antigens. J Exp Med 2002. 196:1099-1104. [DOD] [CrossRef]
- Calderon B, Suri A, Miller MJ, Unanue ER. Dendritic cells in islets of Langerhans constitutively present beta cell-derived peptides bound to their class II MHC molecules. Proc Natl Acad Sci U S A 2008. 105:6121-6126. [DOD] [CrossRef]
- Mohan JF, Levisetti MG, Calderon B, Herzog JW, Petzold SJ, Unanue ER. Unique autoreactive T cells recognize insulin peptides generated within the islets of Langerhans in autoimmune diabetes. Nat Immunol 2010. 11:350-354. [DOD] [CrossRef]
- Saxena V, Ondr JK, Magnusen AF, Munn DH, Katz JD. The countervailing actions of myeloid and plasmacytoid dendritic cells control autoimmune diabetes in the nonobese diabetic mouse. J Immunol 2007. 179:5041-5053. [DOD]
- Yamanouchi J, Verdaguer J, Han B, Amrani A, Serra P, Santamaria P. Cross-priming of diabetogenic T cells dissociated from CTL-induced shedding of beta cell autoantigens. J Immunol 2003. 171:6900-6909. [DOD]
- Hoglund P, Mintern J, Waltzinger C, Heath W, Benoist C, Mathis D. Initiation of autoimmune diabetes by developmentally regulated presentation of islet cell antigens in the pancreatic lymph nodes. J Exp Med 1999. 189:331-339. [DOD] [CrossRef]
- Kurts C, Heath WR, Carbone FR, Allison J, Miller JF, Kosaka H. Constitutive class I-restricted exogenous presentation of self antigens in vivo. J Exp Med 1996. 184:923-930. [DOD] [CrossRef]
- Mintern JD, Sutherland RM, Lew AM, Shortman K, Carbone FR, Heath WR. Constitutive, but not inflammatory, cross-presentation is disabled in the pancreas of young mice. Eur J Immunol 2002. 32:1044-1051. [DOD] [CrossRef]
- Gagnerault MC, Luan JJ, Lotton C, Lepault F. Pancreatic lymph nodes are required for priming of beta cell reactive T cells in NOD mice. J Exp Med 2002. 196:369-377. [DOD] [CrossRef]
- Regoli M, Bertelli E, Orazioli D, Fonzi L, Bastianini A. Pancreatic lymphatic system in rodents. Anat Rec 2001. 263:155-160. [DOD] [CrossRef]
- Rosmalen JG, Homo-Delarche F, Durant S, Kap M, Leenen PJ, Drexhage HA. Islet abnormalities associated with an early influx of dendritic cells and macrophages in NOD and NODscid mice. Lab Invest 2000. 80:769-777. [DOD] [CrossRef]
- Tang Q, Adams JY, Tooley AJ, Bi M, Fife BT, Serra P, Santamaria P, Locksley RM, Krummel MF, Bluestone JA. Visualizing regulatory T cell control of autoimmune responses in nonobese diabetic mice. Nat Immunol 2006. 7:83-92. [DOD] [CrossRef]
- Van Belle TL, Nierkens S, Arens R, von Herrath MG. Interleukin-21 receptor-mediated signals control autoreactive T cell infiltration in pancreatic islets. Immunity 2012. 36:1060-1072. [DOD] [CrossRef]
- Yin N, Xu J, Ginhoux F, Randolph GJ, Merad M, Ding Y, Bromberg JS. Functional specialization of islet dendritic cell subsets. J Immunol 2012. 188:4921-4930. [DOD] [CrossRef]
- Katz JD, Ondr JK, Opoka RJ, Garcia Z, Janssen EM. Cutting edge: merocytic dendritic cells break T cell tolerance to beta cell antigens in nonobese diabetic mouse diabetes. J Immunol 2010. 185:1999-2003. [DOD] [CrossRef]
- Kurts C, Sutherland RM, Davey G, Li M, Lew AM, Blanas E, Carbone FR, Miller JF, Heath WR. CD8 T cell ignorance or tolerance to islet antigens depends on antigen dose. Proc Natl Acad Sci U S A 1999. 96:12703-12707. [DOD] [CrossRef]
- O'Keeffe M, Brodnicki TC, Fancke B, Vremec D, Morahan G, Maraskovsky E, Steptoe R, Harrison LC, Shortman K. Fms-like tyrosine kinase 3 ligand administration overcomes a genetically determined dendritic cell deficiency in NOD mice and protects against diabetes development. Int Immunol 2005. 17:307-314. [DOD] [CrossRef]
- Vasquez AC, Feili-Hariri M, Tan RJ, Morel PA. Qualitative and quantitative abnormalities in splenic dendritic cell populations in NOD mice. Clin Exp Immunol 2004. 135:209-218. [DOD] [CrossRef]
- Kurts C, Carbone FR, Barnden M, Blanas E, Allison J, Heath WR, Miller JF. CD4+ T cell help impairs CD8+ T cell deletion induced by cross-presentation of self-antigens and favors autoimmunity. J Exp Med 1997. 186:2057-2062. [DOD] [CrossRef]
- Stephens LA, Kay TW. Pancreatic expression of B7 co-stimulatory molecules in the non-obese diabetic mouse. Int Immunol 1995. 7:1885-1895. [DOD] [CrossRef]
- Stadinski BD, Delong T, Reisdorph N, Reisdorph R, Powell RL, Armstrong M, Piganelli JD, Barbour G, Bradley B, Crawford F, et al. Chromogranin A is an autoantigen in type 1 diabetes. Nat Immunol 2010. 11:225-231. [DOD] [CrossRef]
- Krishnamurthy B, Dudek NL, McKenzie MD, Purcell AW, Brooks AG, Gellert S, Colman PG, Harrison LC, Lew AM, Thomas HE, Kay TW. Responses against islet antigens in NOD mice are prevented by tolerance to proinsulin but not IGRP. J Clin Invest 2006. 116:3258-3265. [DOD] [CrossRef]
- Jaeckel E, Klein L, Martin-Orozco N, von Boehmer H. Normal incidence of diabetes in NOD mice tolerant to glutamic acid decarboxylase. J Exp Med 2003. 197:1635-1644. [DOD] [CrossRef]
- French MB, Allison J, Cram DS, Thomas HE, Dempsey-Collier M, Silva A, Georgiou HM, Kay TW, Harrison LC, Lew AM. Transgenic expression of mouse proinsulin II prevents diabetes in nonobese diabetic mice. Diabetes 1997. 46:34-39. [DOD] [CrossRef]
- DiLorenzo TP. Multiple antigens versus single major antigen in type 1 diabetes: arguing for multiple antigens. Diabetes Metab Res Rev 2011. 27:778-783. [DOD] [CrossRef]
- Winer S, Tsui H, Lau A, Song A, Li X, Cheung RK, Sampson A, Afifiyan F, Elford A, Jackowski G, Becker DJ, Santamaria P, Ohashi P, Dosch HM. Autoimmune islet destruction in spontaneous type 1 diabetes is not beta-cell exclusive. Nat Med 2003. 9:198-205. [DOD] [CrossRef]
- Jaeckel E, Lipes MA, von Boehmer H. Recessive tolerance to preproinsulin 2 reduces but does not abolish type 1 diabetes. Nat Immunol 2004. 5:1028-1035. [DOD] [CrossRef]
- Moriyama H, Abiru N, Paronen J, Sikora K, Liu E, Miao D, Devendra D, Beilke J, Gianani R, Gill RG, Eisenbarth GS. Evidence for a primary islet autoantigen (preproinsulin 1) for insulitis and diabetes in the nonobese diabetic mouse. Proc Natl Acad Sci U S A 2003. 100:10376-10381. [DOD] [CrossRef]
- Thebault-Baumont K, Dubois-Laforgue D, Krief P, Briand JP, Halbout P, Vallon-Geoffroy K, Morin J, Laloux V, Lehuen A, Carel JC, et al. Acceleration of type 1 diabetes mellitus in proinsulin 2-deficient NOD mice. J Clin Invest 2003. 111:851-857. [DOD]
- Nakayama M, Abiru N, Moriyama H, Babaya N, Liu E, Miao D, Yu L, Wegmann DR, Hutton JC, Elliott JF, Eisenbarth GS. Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice. Nature 2005. 435:220-223. [DOD] [CrossRef]
- Kubosaki A, Gross S, Miura J, Saeki K, Zhu M, Nakamura S, Hendriks W, Notkins AL. Targeted disruption of the IA-2beta gene causes glucose intolerance and impairs insulin secretion but does not prevent the development of diabetes in NOD mice. Diabetes 2004. 53:1684-1691. [DOD] [CrossRef]
- Yamamoto T, Yamato E, Tashiro F, Sato T, Noso S, Ikegami H, Tamura S, Yanagawa Y, Miyazaki JI. Development of autoimmune diabetes in glutamic acid decarboxylase 65 (GAD65) knockout NOD mice. Diabetologia 2004. 47:221-224. [DOD] [CrossRef]
- Levisetti MG, Suri A, Petzold SJ, Unanue ER. The insulin-specific T cells of nonobese diabetic mice recognize a weak MHC-binding segment in more than one form. J Immunol 2007. 178:6051-6057. [DOD]
- Mohan JF, Petzold SJ, Unanue ER. Register shifting of an insulin peptide-MHC complex allows diabetogenic T cells to escape thymic deletion. J Exp Med 2011. 208:2375-2383. [DOD] [CrossRef]
- Zhang L, Nakayama M, Eisenbarth GS. Insulin as an autoantigen in NOD/human diabetes. Curr Opin Immunol 2008. 20:111-118. [DOD] [CrossRef]
- Narendran P, Mannering SI, Harrison LC. Proinsulin-a pathogenic autoantigen in type 1 diabetes. Autoimmun Rev 2003. 2:204-210. [DOD] [CrossRef]
- Pugliese A, Zeller M, Fernandez A Jr, Zalcberg LJ, Bartlett RJ, Ricordi C, Pietropaolo M, Eisenbarth GS, Bennett ST, Patel DD. The insulin gene is transcribed in the human thymus and transcription levels correlated with allelic variation at the INS VNTR-IDDM2 susceptibility locus for type 1 diabetes. Nat Genet 1997. 15:293-297. [DOD] [CrossRef]
- Bennett ST, Lucassen AM, Gough SC, Powell EE, Undlien DE, Pritchard LE, Merriman ME, Kawaguchi Y, Dronsfield MJ, Pociot F, et al. Susceptibility to human type 1 diabetes at IDDM2 is determined by tandem repeat variation at the insulin gene minisatellite locus. Nat Genet 1995. 9:284-292. [DOD] [CrossRef]
- Mallone R, Martinuzzi E, Blancou P, Novelli G, Afonso G, Dolz M, Bruno G, Chaillous L, Chatenoud L, Bach JM, van Endert P. CD8+ T-cell responses identify beta-cell autoimmunity in human type 1 diabetes. Diabetes 2007. 56:613-621. [DOD] [CrossRef]
- Skowera A, Ellis RJ, Varela-Calvino R, Arif S, Huang GC, Van-Krinks C, Zaremba A, Rackham C, Allen JS, Tree TI, et al. CTLs are targeted to kill beta cells in patients with type 1 diabetes through recognition of a glucose-regulated preproinsulin epitope. J Clin Invest 2008. 118:3390-3402. [DOD]
- Bulek AM, Cole DK, Skowera A, Dolton G, Gras S, Madura F, Fuller A, Miles JJ, Gostick E, Price DA, et al. Structural basis for the killing of human beta cells by CD8(+) T cells in type 1 diabetes. Nat Immunol 2012. 13:283-289. [DOD] [CrossRef]
- Kronenberg D, Knight RR, Estorninho M, Ellis RJ, Kester MG, de Ru A, Eichmann M, Huang GC, Powrie J, Dayan CM, et al. Circulating preproinsulin signal peptide-specific CD8 T cells restricted by the susceptibility molecule HLA-A24 are expanded at onset of type 1 diabetes and kill beta-cells. Diabetes 2012. 61:1752-1759. [DOD] [CrossRef]
- Mannering SI, Harrison LC, Williamson NA, Morris JS, Thearle DJ, Jensen KP, Kay TW, Rossjohn J, Falk BA, Nepom GT, Purcell AW. The insulin A-chain epitope recognized by human T cells is posttranslationally modified. J Exp Med 2005. 202:1191-1197. [DOD] [CrossRef]
- Mannering SI, Pang SH, Williamson NA, Naselli G, Reynolds EC, O'Brien-Simpson NM, Purcell AW, Harrison LC. The A-chain of insulin is a hot-spot for CD4+ T cell epitopes in human type 1 diabetes. Clin Exp Immunol 2009. 156:226-231. [DOD] [CrossRef]
- Roep BO, Peakman M. Antigen targets of type 1 diabetes autoimmunity. Cold Spring Harb Perspect Med 2012. 2:a007781. [DOD] [CrossRef]
- Delong T, Baker RL, He J, Barbour G, Bradley B, Haskins K. Diabetogenic T-cell clones recognize an altered Peptide of chromogranin a. Diabetes. 61:3239-3246. [DOD] [CrossRef]
- Calderon B, Carrero JA, Miller MJ, Unanue ER. Cellular and molecular events in the localization of diabetogenic T cells to islets of Langerhans. Proc Natl Acad Sci U S A 2011. 108:1561-1566. [DOD] [CrossRef]
- Calderon B, Carrero JA, Miller MJ, Unanue ER. Entry of diabetogenic T cells into islets induces changes that lead to amplification of the cellular response. Proc Natl Acad Sci U S A 2011. 108:1567-1572. [DOD] [CrossRef]
- Savinov AY, Wong FS, Stonebraker AC, Chervonsky AV. Presentation of antigen by endothelial cells and chemoattraction are required for homing of insulin-specific CD8+ T cells. J Exp Med 2003. 197:643-656. [DOD] [CrossRef]
- Lennon GP, Bettini M, Burton AR, Vincent E, Arnold PY, Santamaria P, Vignali DA. T cell islet accumulation in type 1 diabetes is a tightly regulated, cell-autonomous event. Immunity 2009. 31:643-653. [DOD] [CrossRef]
- Wang J, Tsai S, Shameli A, Yamanouchi J, Alkemade G, Santamaria P. In situ recognition of autoantigen as an essential gatekeeper in autoimmune CD8+ T cell inflammation. Proc Natl Acad Sci U S A 2010. 107:9317-9322. [DOD] [CrossRef]
- Frigerio S, Junt T, Lu B, Gerard C, Zumsteg U, Hollander GA, Piali L. Beta cells are responsible for CXCR3-mediated T-cell infiltration in insulitis. Nat Med 2002. 8:1414-1420. [DOD] [CrossRef]
- Weiss L, Slavin S, Reich S, Cohen P, Shuster S, Stern R, Kaganovsky E, Okon E, Rubinstein AM, Naor D. Induction of resistance to diabetes in non-obese diabetic mice by targeting CD44 with a specific monoclonal antibody. Proc Natl Acad Sci U S A 2000. 97:285-290. [DOD] [CrossRef]
- Savinov AY, Burn P. Interference with islet-specific homing of autoreactive T cells: an emerging therapeutic strategy for type 1 diabetes. Drug Discov Today 2010. 15:531-539. [DOD] [CrossRef]
- Dahlen E, Dawe K, Ohlsson L, Hedlund G. Dendritic cells and macrophages are the first and major producers of TNF-alpha in pancreatic islets in the nonobese diabetic mouse. J Immunol 1998. 160:3585-3593. [DOD]
- Jansen A, Homo-Delarche F, Hooijkaas H, Leenen PJ, Dardenne M, Drexhage HA. Immunohistochemical characterization of monocytes-macrophages and dendritic cells involved in the initiation of the insulitis and beta-cell destruction in NOD mice. Diabetes 1994. 43:667-675. [DOD] [CrossRef]
- Yang Y, Charlton B, Shimada A, Dal Canto R, Fathman CG. Monoclonal T cells identified in early NOD islet infiltrates. Immunity 1996. 4:189-194. [DOD] [CrossRef]
- Bonner-Weir S. The microvasculature of the pancreas, with emphasis on that of the islets of langerhans. Anatomy and funtional implications. In: Vay Liang W. (Ed.) The pancreas: biology, pathobiology, and disease. Second Edition, Raven Press, New York 1993, pp 759-768. [DOD]
- Irving-Rodgers HF, Ziolkowski AF, Parish CR, Sado Y, Ninomiya Y, Simeonovic CJ, Rodgers RJ. Molecular composition of the peri-islet basement membrane in NOD mice: a barrier against destructive insulitis. Diabetologia 2008. 51:1680-1688. [DOD] [CrossRef]
- Ziolkowski AF, Popp SK, Freeman C, Parish CR, Simeonovic CJ. Heparan sulfate and heparanase play key roles in mouse beta cell survival and autoimmune diabetes. J Clin Invest 2012. 122:132-141. [DOD] [CrossRef]
- Coppieters KT, von Herrath MG. Histopathology of type 1 diabetes: old paradigms and new insights. Rev Diabet Stud 2009. 6(2):85-96. [DOD] [CrossRef]
- Bottazzo GF, Dean BM, McNally JM, MacKay EH, Swift PG, Gamble DR. In situ characterization of autoimmune phenomena and expression of HLA molecules in the pancreas in diabetic insulitis. N Engl J Med 1985. 313:353-360. [DOD] [CrossRef]
- Foulis AK, Liddle CN, Farquharson MA, Richmond JA, Weir RS. The histopathology of the pancreas in type 1 (insulin-dependent) diabetes mellitus: a 25-year review of deaths in patients under 20 years of age in the United Kingdom. Diabetologia 1986. 29:267-274. [DOD] [CrossRef]
- Itoh N, Hanafusa T, Miyazaki A, Miyagawa J, Yamagata K, Yamamoto K, Waguri M, Imagawa A, Tamura S, Inada M, et al. Mononuclear cell infiltration and its relation to the expression of major histocompatibility complex antigens and adhesion molecules in pancreas biopsy specimens from newly diagnosed insulin-dependent diabetes mellitus patients. J Clin Invest 1993. 92:2313-2322. [DOD] [CrossRef]
- In't Veld P, Lievens D, De Grijse J, Ling Z, Van der Auwera B, Pipeleers-Marichal M, Gorus F, Pipeleers D. Screening for insulitis in adult autoantibody-positive organ donors. Diabetes 2007. 56:2400-2404. [DOD] [CrossRef]
- Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol 2009. 155:173-181. [DOD] [CrossRef]
- Keenan HA, Sun JK, Levine J, Doria A, Aiello LP, Eisenbarth G, Bonner-Weir S, King GL. Residual insulin production and pancreatic ss-cell turnover after 50 years of diabetes: Joslin Medalist Study. Diabetes 2010. 59:2846-2853. [DOD] [CrossRef]
- Coppieters KT, Dotta F, Amirian N, Campbell PD, Kay TW, Atkinson MA, Roep BO, von Herrath MG. Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J Exp Med 2012. 209:51-60. [DOD] [CrossRef]
- Katz J, Benoist C, Mathis D. Major histocompatibility complex class I molecules are required for the development of insulitis in non-obese diabetic mice. Eur J Immunol 1993. 23:3358-3360. [DOD] [CrossRef]
- Serreze DV, Leiter EH, Christianson GJ, Greiner D, Roopenian DC. Major histocompatibility complex class I-deficient NOD-B2mnull mice are diabetes and insulitis resistant. Diabetes 1994. 43:505-509. [DOD] [CrossRef]
- Sumida T, Furukawa M, Sakamoto A, Namekawa T, Maeda T, Zijlstra M, Iwamoto I, Koike T, Yoshida S, et al. Prevention of insulitis and diabetes in beta 2-microglobulin-deficient non-obese diabetic mice. Int Immunol 1994. 6:1445-1449. [DOD] [CrossRef]
- Wicker LS, Leiter EH, Todd JA, Renjilian RJ, Peterson E, Fischer PA, Podolin PL, Zijlstra M, Jaenisch R, Peterson LB. Beta 2-microglobulin-deficient NOD mice do not develop insulitis or diabetes. Diabetes 1994. 43:500-504. [DOD] [CrossRef]
- Kay TW, Parker JL, Stephens LA, Thomas HE, Allison J. RIP-beta 2-microglobulin transgene expression restores insulitis, but not diabetes, in beta 2-microglobulin null nonobese diabetic mice. J Immunol 1996. 157:3688-3693. [DOD]
- Chong MM, Chen Y, Darwiche R, Dudek NL, Irawaty W, Santamaria P, Allison J, Kay TW, Thomas HE. Suppressor of cytokine signaling-1 overexpression protects pancreatic beta cells from CD8+ T cell-mediated autoimmune destruction. J Immunol 2004. 172:5714-5721. [DOD]
- Dudek NL, Thomas HE, Mariana L, Sutherland RM, Allison J, Estella E, Angstetra E, Trapani JA, Santamaria P, Lew AM, Kay TW. Cytotoxic T-cells from T-cell receptor transgenic NOD8.3 mice destroy beta-cells via the perforin and Fas pathways. Diabetes 2006. 55:2412-2418. [DOD] [CrossRef]
- Hamilton-Williams EE, Palmer SE, Charlton B, Slattery RM. Beta cell MHC class I is a late requirement for diabetes. Proc Natl Acad Sci U S A 2003. 100:6688-6693. [DOD] [CrossRef]
- Coppieters K, Amirian N, von Herrath M. Intravital imaging of CTLs killing islet cells in diabetic mice. J Clin Invest 2012. 122:119-131. [DOD] [CrossRef]
- Kagi D, Odermatt B, Seiler P, Zinkernagel RM, Mak TW, Hengartner H. Reduced incidence and delayed onset of diabetes in perforin-deficient nonobese diabetic mice. J Exp Med 1997. 186:989-997. [DOD] [CrossRef]
- Amrani A, Verdaguer J, Anderson B, Utsugi T, Bou S, Santamaria P. Perforin-independent beta-cell destruction by diabetogenic CD8(+) T lymphocytes in transgenic nonobese diabetic mice. J Clin Invest 1999. 103:1201-1209. [DOD] [CrossRef]
- Cantor J, Haskins K. Effector function of diabetogenic CD4 Th1 T cell clones: a central role for TNF-alpha. J Immunol 2005. 175:7738-7745. [DOD]
- Amrani A, Verdaguer J, Thiessen S, Bou S, Santamaria P. IL-1alpha, IL-1beta, and IFN-gamma mark beta cells for Fas-dependent destruction by diabetogenic CD4(+) T lymphocytes. J Clin Invest 2000. 105:459-468. [DOD] [CrossRef]
- Estella E, McKenzie MD, Catterall T, Sutton VR, Bird PI, Trapani JA, Kay TW, Thomas HE. Granzyme B-mediated death of pancreatic beta-cells requires the proapoptotic BH3-only molecule bid. Diabetes 2006. 55:2212-2219. [DOD] [CrossRef]
- Mollah ZU, Graham KL, Krishnamurthy B, Trivedi P, Brodnicki TC, Trapani JA, Kay TW, Thomas HE. Granzyme B is dispensable in the development of diabetes in non-obese diabetic mice. PLoS One 2012. 7:e40357. [DOD] [CrossRef]
- Darwiche R, Chong MM, Santamaria P, Thomas HE, Kay TW. Fas is detectable on beta cells in accelerated, but not spontaneous, diabetes in nonobese diabetic mice. J Immunol 2003. 170:6292-6297. [DOD]
- Chervonsky AV, Wang Y, Wong FS, Visintin I, Flavell RA, Janeway CA Jr, Matis LA. The role of Fas in autoimmune diabetes. Cell 1997. 89:17-24. [DOD] [CrossRef]
- Allison J, Strasser A. Mechanisms of beta cell death in diabetes: a minor role for CD95. Proc Natl Acad Sci U S A 1998. 95:13818-13822. [DOD] [CrossRef]
- Allison J, Thomas HE, Catterall T, Kay TW, Strasser A. Transgenic expression of dominant-negative Fas-associated death domain protein in beta cells protects against Fas ligand-induced apoptosis and reduces spontaneous diabetes in nonobese diabetic mice. J Immunol 2005. 175:293-301. [DOD]
- Savinov AY, Tcherepanov A, Green EA, Flavell RA, Chervonsky AV. Contribution of Fas to diabetes development. Proc Natl Acad Sci U S A 2003. 100:628-632. [DOD] [CrossRef]
- Angstetra E, Graham KL, Emmett S, Dudek NL, Darwiche R, Ayala-Perez R, Allison J, Santamaria P, Kay TW, Thomas HE. In vivo effects of cytokines on pancreatic beta-cells in models of type I diabetes dependent on CD4(+) T lymphocytes. Immunol Cell Biol 2009. 87:178-185. [DOD] [CrossRef]
- Apostolou I, Hao Z, Rajewsky K, von Boehmer H. Effective destruction of Fas-deficient insulin-producing beta cells in type 1 diabetes. J Exp Med 2003. 198:1103-1106. [DOD] [CrossRef]
- Mollah ZU, Wali J, McKenzie MD, Krishnamurthy B, Graham KL, Fynch S, Szanyi J, Santamaria P, Brodnicki T, Allison J, et al. The pro-apoptotic BH3-only protein Bid is dispensable for development of insulitis and diabetes in the non-obese diabetic mouse. Apoptosis 2011. 16:822-830. [DOD] [CrossRef]
- McKenzie MD, Carrington EM, Kaufmann T, Strasser A, Huang DC, Kay TW, Allison J, Thomas HE. Proapoptotic BH3-only protein Bid is essential for death receptor-induced apoptosis of pancreatic beta-cells. Diabetes 2008. 57:1284-1292. [DOD] [CrossRef]
- Knight RR, Kronenberg D, Zhao M, Huang GC, Eichmann M, Bulek A, Wooldridge L, Cole DK, Sewell AK, Peakman M, Skowera A. Human beta-cell killing by autoreactive preproinsulin-specific CD8 T cells is predominantly granule-mediated with the potency dependent upon T-cell receptor avidity. Diabetes 2012. In press. [DOD]
- Foulis AK, Farquharson MA, Hardman R. Aberrant expression of class II major histocompatibility complex molecules by B cells and hyperexpression of class I major histocompatibility complex molecules by insulin containing islets in type 1 (insulin-dependent) diabetes mellitus. Diabetologia 1987. 30:333-343. [DOD] [CrossRef]
- Campbell PD, Estella E, Dudek NL, Jhala G, Thomas HE, Kay TW, Mannering SI. Cytotoxic T-lymphocyte-mediated killing of human pancreatic islet cells in vitro. Hum Immunol 2008. 69:543-551. [DOD] [CrossRef]
- Eizirik DL, Mandrup-Poulsen T. A choice of death--the signal-transduction of immune-mediated beta-cell apoptosis. Diabetologia 2001. 44:2115-2133. [DOD] [CrossRef]
- Barthson J, Germano CM, Moore F, Maida A, Drucker DJ, Marchetti P, Gysemans C, Mathieu C, Nunez G, Jurisicova A, at al. Cytokines tumor necrosis factor-alpha and interferon-gamma induce pancreatic beta-cell apoptosis through STAT1-mediated Bim protein activation. J Biol Chem 2011. 286:39632-39643. [DOD] [CrossRef]
- Thomas HE, Irawaty W, Darwiche R, Brodnicki TC, Santamaria P, Allison J, Kay TW. IL-1 receptor deficiency slows progression to diabetes in the NOD mouse. Diabetes 2004. 53:113-121. [DOD] [CrossRef]
- Thomas HE, Graham KL, Angstetra E, McKenzie MD, Dudek NL, Kay TW. Interferon signalling in pancreatic beta cells. Front Biosci 2009. 14:644-656. [DOD] [CrossRef]
- O'Sullivan BJ, Thomas HE, Pai S, Santamaria P, Iwakura Y, Steptoe RJ, Kay TW, Thomas R. IL-1 beta breaks tolerance through expansion of CD25+ effector T cells. J Immunol 2006. 176:7278-7287. [DOD]
- Kagi D, Ho A, Odermatt B, Zakarian A, Ohashi PS, Mak TW. TNF receptor 1-dependent beta cell toxicity as an effector pathway in autoimmune diabetes. J Immunol 1999. 162:4598-4605. [DOD]
- Chee J, Angstetra E, Mariana L, Graham KL, Carrington EM, Bluethmann H, Santamaria P, Allison J, Kay TW, Krishnamurthy B, Thomas HE. TNF receptor 1 deficiency increases regulatory T cell function in nonobese diabetic mice. J Immunol 2011. 187:1702-1712. [DOD] [CrossRef]
- Pakala SV, Chivetta M, Kelly CB, Katz JD. In autoimmune diabetes the transition from benign to pernicious insulitis requires an islet cell response to tumor necrosis factor alpha. J Exp Med 1999. 189:1053-1062. [DOD] [CrossRef]
- Gianani R, Verge CF, Moromisato-Gianani RI, Yu L, Zhang YJ, Pugliese A, Eisenbarth GS. Limited loss of tolerance to islet autoantigens in ICA+ first degree relatives of patients with type I diabetes expressing the HLA dqb*10602 allele. J Autoimmun 1996. 9:423-425. [DOD] [CrossRef]
- Sheehy MJ, Scharf SJ, Rowe JR, Neme de Gimenez MH, Meske LM, Erlich HA, Nepom BS. A diabetes-susceptible HLA haplotype is best defined by a combination of HLA-DR and -DQ alleles. J Clin Invest 1989. 83:830-835. [DOD] [CrossRef]
- Onengut-Gumuscu S, Concannon P. Mapping genes for autoimmunity in humans: type 1 diabetes as a model. Immunol Rev 2002. 190:182-194. [DOD] [CrossRef]
- Mannering SI, Wong FS, Durinovic-Bello I, Brooks-Worrell B, Tree TI, Cilio CM, Schloot NC, Mallone R. Current approaches to measuring human islet-antigen specific T cell function in type 1 diabetes. Clin Exp Immunol 2010. 162:197-209. [DOD] [CrossRef]
- De Berardinis P, Londei M, Kahan M, Balsano F, Kontiainen S, Gale EA, Bottazzo GF, Feldmann M. The majority of the activated T cells in the blood of insulin-dependent diabetes mellitus (IDDM) patients are CD4+. Clin Exp Immunol 1988. 73:255-259. [DOD]
- Al-Sakkaf L, Pozzilli P, Tarn AC, Schwarz G, Gale EA, Bottazzo GF. Persistent reduction of CD4/CD8 lymphocyte ratio and cell activation before the onset of type 1 (insulin-dependent) diabetes. Diabetologia 1989. 32:322-325. [DOD] [CrossRef]
- Marwaha AK, Crome SQ, Panagiotopoulos C, Berg KB, Qin H, Ouyang Q, Xu L, Priatel JJ, Levings MK, Tan R. Cutting edge: Increased IL-17-secreting T cells in children with new-onset type 1 diabetes. J Immunol 2010. 185:3814-3818. [DOD] [CrossRef]
- Cooke A. Th17 cells in inflammatory conditions. Rev Diabet Stud 2006. 3(2):72-75. [DOD] [CrossRef]
- Ferraro A, Socci C, Stabilini A, Valle A, Monti P, Piemonti L, Nano R, Olek S, Maffi P, Scavini M, et al. Expansion of Th17 cells and functional defects in T regulatory cells are key features of the pancreatic lymph nodes in patients with type 1 diabetes. Diabetes 2011. 60:2903-2913. [DOD] [CrossRef]
- Arif S, Moore F, Marks K, Bouckenooghe T, Dayan CM, Planas R, Vives-Pi M, Powrie J, Tree T, Marchetti P, et al. Peripheral and islet interleukin-17 pathway activation characterizes human autoimmune diabetes and promotes cytokine-mediated beta-cell death. Diabetes 2011. 60:2112-2119. [DOD] [CrossRef]
- Brusko T, Wasserfall C, McGrail K, Schatz R, Viener HL, Schatz D, Haller M, Rockell J, Gottlieb P, Clare-Salzler M, Atkinson M. No alterations in the frequency of FOXP3+ regulatory T-cells in type 1 diabetes. Diabetes 2007. 56:604-612. [DOD] [CrossRef]
- Lindley S, Dayan CM, Bishop A, Roep BO, Peakman M, Tree TI. Defective suppressor function in CD4(+)CD25(+) T-cells from patients with type 1 diabetes. Diabetes 2005. 54:92-99. [DOD] [CrossRef]
- Martin AP, Rankin S, Pitchford S, Charo IF, Furtado GC, Lira SA. Increased expression of CCL2 in insulin-producing cells of transgenic mice promotes mobilization of myeloid cells from the bone marrow, marked insulitis, and diabetes. Diabetes 2008. 57:3025-3033. [DOD] [CrossRef]
- Underhill DM, Goodridge HS. Information processing during phagocytosis. Nat Rev Immunol 2012. 12:492-502. [DOD] [CrossRef]
- Angstetra E, Graham KL, Zhao Y, Irvin AE, Elkerbout L, Santamaria P, Slattery RM, Kay TW, Thomas HE. An indirect role for NK cells in a CD4(+) T-cell-dependent mouse model of type I diabetes. Immunol Cell Biol 2011. 90:243-247. [DOD] [CrossRef]
- Feuerer M, Shen Y, Littman DR, Benoist C, Mathis D. How punctual ablation of regulatory T cells unleashes an autoimmune lesion within the pancreatic islets. Immunity 2009. 31:654-664. [DOD] [CrossRef]
- Tang Q, Adams JY, Penaranda C, Melli K, Piaggio E, Sgouroudis E, Piccirillo CA, Salomon BL, Bluestone JA. Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity 2008. 28:687-697. [DOD] [CrossRef]
- Yamanouchi J, Rainbow D, Serra P, Howlett S, Hunter K, Garner VE, Gonzalez-Munoz A, Clark J, Veijola R, Cubbon R, et al. Interleukin-2 gene variation impairs regulatory T cell function and causes autoimmunity. Nat Genet 2007. 39:329-337. [DOD] [CrossRef]
- You S, Belghith M, Cobbold S, Alyanakian MA, Gouarin C, Barriot S, Garcia C, Waldmann H, Bach JF, Chatenoud L. Autoimmune diabetes onset results from qualitative rather than quantitative age-dependent changes in pathogenic T-cells. Diabetes 2005. 54:1415-1422. [DOD] [CrossRef]
- Long SA, Cerosaletti K, Bollyky PL, Tatum M, Shilling H, Zhang S, Zhang ZY, Pihoker C, Sanda S, Greenbaum C, Buckner JH. Defects in IL-2R signaling contribute to diminished maintenance of FOXP3 expression in CD4(+)CD25(+) regulatory T-cells of type 1 diabetic subjects. Diabetes 2010. 59:407-415. [DOD] [CrossRef]
- McGuire HM, Vogelzang A, Hill N, Flodstrom-Tullberg M, Sprent J, King C. Loss of parity between IL-2 and IL-21 in the NOD Idd3 locus. Proc Natl Acad Sci U S A 2009. 106:19438-19443. [DOD] [CrossRef]
- McGuire HM, Walters S, Vogelzang A, Lee CM, Webster KE, Sprent J, Christ D, Grey S, King C. Interleukin-21 is critically required in autoimmune and allogeneic responses to islet tissue in murine models. Diabetes 2011. 60:867-875. [DOD] [CrossRef]
- Spolski R, Kashyap M, Robinson C, Yu Z, Leonard WJ. IL-21 signaling is critical for the development of type I diabetes in the NOD mouse. Proc Natl Acad Sci U S A 2008. 105:14028-14033. [DOD] [CrossRef]
- Sutherland AP, Van Belle T, Wurster AL, Suto A, Michaud M, Zhang D, Grusby MJ, von Herrath M. Interleukin-21 is required for the development of type 1 diabetes in NOD mice. Diabetes 2009. 58:1144-1155. [DOD] [CrossRef]
- Lee LF, Logronio K, Tu GH, Zhai W, Ni I, Mei L, Dilley J, Yu J, Rajpal A, Brown C, et al. Anti-IL-7 receptor-alpha reverses established type 1 diabetes in nonobese diabetic mice by modulating effector T-cell function. Proc Natl Acad Sci U S A 2012. 109:12674-12679. [DOD] [CrossRef]
- Penaranda C, Kuswanto W, Hofmann J, Kenefeck R, Narendran P, Walker LS, Bluestone JA, Abbas AK, Dooms H. IL-7 receptor blockade reverses autoimmune diabetes by promoting inhibition of effector/memory T cells. Proc Natl Acad Sci U S A 2012. 109:12668-12673. [DOD] [CrossRef]
- Fife BT, Guleria I, Gubbels Bupp M, Eagar TN, Tang Q, Bour-Jordan H, Yagita H, Azuma M, Sayegh MH, Bluestone JA. Insulin-induced remission in new-onset NOD mice is maintained by the PD-1-PD-L1 pathway. J Exp Med 2006. 203:2737-2747. [DOD] [CrossRef]
- Fife BT, Pauken KE, Eagar TN, Obu T, Wu J, Tang Q, Azuma M, Krummel MF, Bluestone JA. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat Immunol 2009. 10:1185-1192. [DOD] [CrossRef]
- Brodie GM, Wallberg M, Santamaria P, Wong FS, Green EA. B-cells promote intra-islet CD8+ cytotoxic T-cell survival to enhance type 1 diabetes. Diabetes 2008. 57:909-917. [DOD] [CrossRef]
- Penaranda C, Tang Q, Ruddle NH, Bluestone JA. Prevention of diabetes by FTY720-mediated stabilization of peri-islet tertiary lymphoid organs. Diabetes 2010. 59:1461-1468. [DOD] [CrossRef]
- Graham KL, Krishnamurthy B, Fynch S, Ayala-Perez R, Slattery RM, Santamaria P, Thomas HE, Kay TW. Intra-islet proliferation of cytotoxic T lymphocytes contributes to insulitis progression. Eur J Immunol 2012. 42:1717-1722. [DOD] [CrossRef]
- Graham KL, Krishnamurthy B, Fynch S, Mollah ZU, Slattery R, Santamaria P, Kay TW, Thomas HE. Autoreactive cytotoxic T lymphocytes acquire higher expression of cytotoxic effector markers in the islets of NOD mice after priming in pancreatic lymph nodes. Am J Pathol 2011. 178:2716-2725. [DOD] [CrossRef]
- Danke NA, Yang J, Greenbaum C, Kwok WW. Comparative study of GAD65-specific CD4+ T cells in healthy and type 1 diabetic subjects. J Autoimmun 2005. 25:303-311. [DOD] [CrossRef]
- Trudeau JD, Kelly-Smith C, Verchere CB, Elliott JF, Dutz JP, Finegood DT, Santamaria P, Tan R. Prediction of spontaneous autoimmune diabetes in NOD mice by quantification of autoreactive T cells in peripheral blood. J Clin Invest 2003. 111:217-223. [DOD]
This article has been cited by other articles:

|
Bringing the human pancreas into focus: new paradigms for the understanding of Type 1 diabetes
Morgan NG
Diabet Med 2017. 34(7):879-886
|
|

|
Repurposed JAK1/JAK2 Inhibitor Reverses Established Autoimmune Insulitis in NOD Mice
Trivedi PM, Graham KL, Scott NA, Jenkins MR, Majaw S, Sutherland RM, Fynch S, Lew AM, Burns CJ, Krishnamurthy B, Brodnicki TC, Mannering SI, Kay TW, Thomas HE
Diabetes. 2017. 66(6):1650-1660
|
|

|
Emerging role of Hippo signalling in pancreatic biology: YAP re-expression and plausible link to islet cell apoptosis and replication
Sharma A, Yerra VG, Kumar A
Biochimie 2017. 133:56-65
|
|

|
High-density lipoprotein immunomodulates the functional activities of macrophage and cytokines produced during ex vivo macrophage-CD4+ T cell crosstalk at the recent-onset human type 1 diabetes
Benghalem I, Meziane W, Hadjidj Z, Ysmail-Dahlouk L, Belamri A, Mouhadjer K, Aribi M
Cytokine 2017. 96:59-70
|
|

|
Development of the Nonobese Diabetic Mouse and Contribution of Animal Models for Understanding Type 1 Diabetes
Mullen Y
Pancreas 2017. 46(4):455-466
|
|
|
Isolation and Culture of the Islets of Langerhans from Mouse Pancreas
Graham KL, Fynch S, Papas EG, Tan C, Kay TW, Thomas HE
Bio-protocol 2016. 6(12):e1840
|
|

|
Association between telomere length and diabetes mellitus: A meta-analysis
Wang J, Dong X, Cao L, Sun Y, Qiu Y, Zhang Y, Cao R, Covasa M, Zhong L
J Int Med Res 2016. 44(6):1156-1173
|
|

|
Perforin facilitates beta cell killing and regulates autoreactive CD8+ T-cell responses to antigen in mouse models of type 1 diabetes
Trivedi P, Graham KL, Krishnamurthy B, Fynch S, Slattery RM, Kay TW, Thomas HE
Immunol Cell Biol 2016. 94(4):334-341
|
|

|
Human islet cells are killed by BID-independent mechanisms in response to FAS ligand
Joglekar MV, Trivedi PM, Kay TW, Hawthorne WJ, O'Connell PJ, Jenkins AJ, Hardikar AA, Thomas HE
Apoptosis 2016. 21(4):379-389
|
|

|
Mouse pancreatic beta cells express MHC class II and stimulate CD4+ T cells to proliferate
Zhao Y, Scott NA, Quah HS, Krishnamurthy B, Bond F, Loudovaris T, Mannering SI, Kay TW, Thomas HE
Eur J Immunol 2015. 45(9):2494-503
|
|

|
Beta-cell destruction and preservation in childhood and adult onset type 1 diabetes
Poudel A, Savari O, Striegel DA, Periwal V, Taxy J, Millis JM, Witkowski P, Atkinson MA, Hara M
Endocrine 2015. In press
|
|

|
Therapeutic potential of umbilical cord blood cells for type 1 diabetes mellitus
He B, Li X, Yu H, Zhou Z
J Diabetes 2015. 7(6):762-773
|
|

|
Autoreactive T cells induce necrosis and not BCL-2-regulated or death receptor-mediated apoptosis or RIPK3-dependent necroptosis of transplanted islets in a mouse model of type 1 diabetes
Zhao Y, Scott NA, Fynch S, Elkerbout L, Wong WW, Mason KD, Strasser A, Huang DC, Kay TW, Thomas HE
Diabetologia 2015. 58(1):140-148
|
|

|
Proinsulin-Specific, HLA-DQ8, and HLA-DQ8-Transdimer-Restricted CD4+ T Cells Infiltrate Islets in Type 1 Diabetes
Pathiraja V, Kuehlich JP, Campbell PD, Krishnamurthy B, Loudovaris T, Coates PT, Brodnicki TC, O'Connell PJ, Kedzierska K, Rodda C, Bergman P, Hill E, Purcell AW, Dudek NL, Thomas HE, Kay TW, Mannering SI
Diabetes 2014. In press
|
|

|
Islet inflammation in human type 1 diabetes mellitus
Morgan NG, Leete P, Foulis AK, Richardson SJ
IUBMB Life 2014. 66(11):723-734
|
|
|


)
)
)
)
)

