Chapter I. Pathogenesis
| Rev Diabet Stud,
2012,
9(4):137-147 |
DOI 10.1900/RDS.2012.9.137 |
Genetic Analysis of Type 1 Diabetes: Embryonic Stem Cells as New Tools to Unlock Biological Mechanisms in Type 1 Diabetes
Nick Holmes, Anne Cooke
Department of Pathology, University of Cambridge, Tennis Court Road, Cambridge CB2 1QP, UK
Address correspondence to: Nick Holmes, e-mail nh106@cam.ac.uk
Manuscript submitted December 28, 2012; resubmitted January 9, 2013; accepted January 10, 2013.
Keywords: type 1 diabetes, embryonic stem cell, nonobese diabetic mouse, NOD, gene, susceptibility, haplotype
Abstract
The nonobese diabetic (NOD) mouse has provided an important animal model for studying the mechanism and genetics of type 1 diabetes over the past 30 years. Arguably, the bio-breeding (BB) rat model may be an even closer phenotypic mimic of the typical human disease. A large number of distinct genetic traits which influence diabetes development have been defined through an extraordinary effort, most conspicuously in the mouse model. However, in both NOD and BB models the lack of availability of robust means for experimental genetic manipulation has restricted our understanding of the mechanisms underlying this spontaneous autoimmune disease. Recent developments in the derivation of embryonic stem (ES) cells have the potential to transform this picture. We argue here that targeting of NOD strain ES cells can bring much needed certainty to our present understanding of the genetics of type 1 diabetes in the NOD mouse. In addition, ES cells can play important roles in the future, in both the NOD mouse and BB rat models, through the generation of new tools to investigate the mechanisms by which genetic variation acts to promote diabetes.
Abbreviations: AB/H - antibody high; AEC - autoimmune exocrinopathy; B2ma - gene controlling β2-microglobulin in NOD mouse; B6 - C57BL/6; B10 - C57BL/10; BB - bio-breeding; C57BL/6 - inbred strain C57 black 6; C57BL/10 - inbred strain C57 black 10; CDK1 - cyclin-dependent kinase 1; DNA - deoxyribonucleic acid; DsRED - Discosoma (sea anemone) red fluorescent protein; EAE - autoimmune encephalomyelitis; ERK - extracellular signal-regulated kinase; ES - embryonic stem; FCGR1 - Fc gamma receptor 1; GSK3 - glycogen synthase kinase 3; H2-Ea - histocompatibility 2, class II antigen E alpha; HLA - human leukocyte antigen; HSV-tk - herpes simplex virus thymidine kinase; Idd - insulin-dependent diabetes; IFNGR1 - interferon gamma receptor 1; IgG - immunoglobulin G; IL-2 - interleukin 2; IL-2RA - IL-2 receptor alpha; iPS cell - induced pluripotent stem cell; Kb - kilobase; LIF - leukemia inhibitory factor; Mb - megabase; MEK - mitogen-activated protein kinase; MHC - major histocompability complex; NON - nonobese nondiabetic; NOD - nonobese diabetic; NOR - nonobese-resistant; NRAMP1 - natural resistance-associated macrophage protein 1; PD-1 - programmed death-1; PTPN22 - protein tyrosine phosphatase non-receptor type 22; Ras-GAP - Ras GTPase-activating protein; RNA - ribonucleic acid; RNAi - RNA interference; shRNA - small hairpin RNA; SLC11 - solute carrier family 11; SNP - single nucleotide polymorphism; T1D - type 1 diabetes; TALEN - transcription activator-like effector nuclease; TCR - T cell receptor; ZFN - zinc finger nucleases
1. NOD congenic mice
In the NOD model, a great deal of effort has been invested in the genetic dissection of disease susceptibility. This has resulted in the description of more than 35 different chromosomal regions, spread over 14 of the 19 mouse autosomes, which influence the development of type 1 diabetes (T1D) in the mouse [1, 2]. The majority of these data have been obtained by painstaking effort to produce a large number of congenic mouse strains of the type in which all but one segment of a single chromosome is derived from the NOD mouse. The non-NOD DNA has been introduced by first intercrossing NOD with a mouse strain which does not spontaneously develop diabetes, and then by serial backcrossing selected animals to NOD, normally for 12 or more generations.
The limits of the introgressed interval can be defined by typing for markers which vary between NOD and the donor strain. Typically, despite the heroic efforts of the investigators, these intervals are several megabases in size and contain several, sometimes dozens, of annotated genes [2]. In many cases, the degree of genetic diversity between the NOD strain and introgressed donor DNA is not fully known. While the C57BL/6 (B6) reference genome is essentially complete and extensive sequence data for NOD exists, annotated sequence coverage of the NOD genome in publically available databases is currently limited. Furthermore, B6 is the congenic donor only in certain strains; C57BL/10SnJ (B10), nonobese nondiabetic (NON), nonobese-resistant (NOR) and, less frequently, many other strains have been used to produce NOD congenics [1]. While B10 has many fewer differences with B6 than NOD [3], these are not all quantified or mapped. There is considerable variation in the degree of sequence diversity between B6 and NOD, but in some places it is high (~1%) [4]. This has helped the precision with which the recombination boundaries of the susceptibility intervals can be defined. However, it means that even regions of 2 Mb typically contain more than a few hundred differences [2] which renders attributing causation to any individual sequence variant impossible.
With the completion of the mouse genome project, it is certainly possible to inspect the susceptibility regions for annotated genes and use knowledge of these genes and their products to make informed guesses about possible candidates. Obviously, in some cases this is easier than in others, but in all cases there must be a degree of uncertainty about the identity of the causative gene and even more so about the identity of the variant or variants which lead to the susceptibility phenotype. It is certainly true that even with a good deal of circumstantial evidence in their favor, hypothetical identification of causative genes may finger the wrong candidate. A good example of this is the Idd3 region on chromosome 3 (see next section).
The study of congenic strains selected for reduced diabetes incidence has been complemented by at least three distinct approaches to probing candidate genes.
2. Correlating genetic, phenotypic, and functional variation
If congenic mouse strains have identified and partially localized a susceptibility/resistance polymorphic trait, an obvious method to identify candidate genes is to examine their known function and to look for specific allelic variation correlating with some functional phenotype which might plausibly be involved in the development of T1D. While the current state of all Idd regions includes multiple genes, some contain deliciously tempting possibilities whose known biology positively invites hypothetical imputation. However, inevitably 'cherry picking' of what appear likely candidates suffers from bias imposed by our ignorance and limited by our imaginations; there is surely a case that imputations have been too immunologically focused and that genes affecting beta-cell intrinsic functions have been too little considered [5].
The first, and still most significant, susceptibility region described was Idd1. This region contains the MHC. It was established early on that the MHC of NOD mice contained an haplotype of linked MHC alleles, shared only with the closely related nonobese-resistant (NOR) strain [6]. Within that haplotype was a single functional MHC class II gene pair with the H2-Ab1g7 allele found only in NOD, NOR, and Biozzi AB/H strains [7]. This class II beta chain had two particularly distinctive amino acids at positions 56 and 57 (HisSer) [6]; position 57 was considered especially significant since human HLA-DQbeta alleles associated with T1D also lacked the Asp residue found at this position in the majority of alleles associated with resistance [8]. Of course, the known function of H-2A was also a strongly supportive factor; polymorphism at this locus affecting peptide selection for antigen presentation, shaping the TCR repertoire, and thus potentially influencing both central and peripheral tolerance. In this case, transgenic mice made on the NOD background showed that additional expression of H2-Ab1g7 genes mutated to express the consensus residue at either position 56 (Pro) or 57 (Asp), either abolished (Pro56) [9] or significantly reduced (Asp57) [10] the incidence of T1D despite the continued expression of the native H2-Ab1g7 allele. Furthermore, it was shown that diabetes onset was dependent on the lack of a functional H2-Ea gene [9]. Even the most cautious commentator would have to say that we are as sure as possible that the H-2Ab1 locus is an important T1D susceptibility locus, and that the 5 nucleotide polymorphisms responsible for the double amino acid change are in all likelihood, at least in part, causative. We can have equal confidence that the 629bp deletion in the H2-Ea gene is also a cause of T1D susceptibility. What we cannot say, however, is that there are no other variants within the NOD MHC significantly affecting diabetes incidence.
The attribution of causality to the H-2Ab1 gene may be correct, but the identification of causative genes and variants within other chromosomal intervals associated with susceptibility or resistance to diabetes has proved fraught with difficulty. This is well illustrated by the history of the Idd3 region. Idd3 was originally described as lying within a large segment of chromosome 3 [11]. However, the disease-associated variants between B6 and NOD on chromosome 3 are now believed to contain no less than six traits that contribute to susceptibility (renamed Idd3, 17, 10, 18.1, 18.3 and 18.4) in NOD mice [2]. Before this dissolution, the gene for the high affinity receptor for IgG (Fcgr1) was the first to be proposed as responsible for the disease linkage [12]. There were sound arguments advanced for this choice. A functional polymorphism was identified, and the minor allele was present in only NOD and another autoimmune-prone strain, Biozzi AB/H (of 22 strains screened). The polymorphism in Fcgr1 was extensive, with 24 nucleotide changes, 19 of them non-synonymous, and resulted in a severe truncation of the cytoplasmic tail of the receptor. The variants affected recycling and/or binding of IgG2a molecules to macrophages. Macrophages were already known to be important in T1D both in NOD and in humans. Nevertheless, subsequent work showed that all susceptibility traits could be separated from the Fcgr1 locus [13].
Following the separation of the chromosome 3 diabetes susceptibility traits into 3 distinct linkage regions, a second plausible candidate was advanced. The now smaller Idd3 region contained the gene for the cytokine IL-2. The size of the potential region was progressively reduced to 780 Kb which still contained IL-2 [14]. A number of significant polymorphisms between NOD and B6 mice were found in the Il2 gene; some were upstream of the gene, while others altered the N-terminal sequence and subsequent glycosylation of the protein. IL-2 was known to be involved in regulating autoimmunity, and IL2RA was a candidate gene in human T1D susceptibility. This led Lyons et al. to examine IL-2 expression levels in Idd3 discordant mice. They found no differences and concluded that the protein differences in IL-2 were most likely responsible for Idd3 [14]. However, a later study by an overlapping group of authors concluded that there were transcriptional differences between strains with Idd3 from different genetic origins, and that the lower level of IL-2 transcription was concordant with susceptibility, whereas protein-altering variants did not correlate in the same comparison [4]. They went on to show that NOD background mice which were heterozygous for an Idd3-containing interval from a 129-derived Il2 knockout had both altered IL-2 levels and disease susceptibility. This later study therefore made the opposite conclusion to the 2000 report, namely that differences in IL-2 expression and not protein-encoding changes were responsible for the Idd3 disease affect.
Nevertheless, while the Yamanouchi study presented a strong argument for polymorphisms upstream of Il2 being responsible, there remains some uncertainty. The discrepancy between the Lyons and the Yamanouchi studies regarding the correlation of IL-2 levels with disease susceptibility illustrates the difficulties in being certain what cell type and/or activation state is the critical population in which to measure expression. The Il21 gene lies within about 100 Kb of Il2 and inside the Idd3 region. There is also compelling evidence that IL-21 to IL-21R interaction is important in T1D [15-18]. The Il2 knockout was made by placing an active neo gene within the third exon of Il2 [19]; to our knowledge no studies have been conducted to show that it does not alter IL-21 expression. Expression of the neo gene is expected to be constitutive and strong, driven by a hybrid HSV-tk promoter and a tandem repeat of the enhancer region from the polyoma mutant PYF441 [20]. Effects on the transcription of neighboring genes have been found in this type of knockout, some at quite long range [21].
3. Introduction of targeted mutations from other genetic backgrounds
A large number of knockout alleles have been backcrossed onto the NOD background; the degree to which these NOD knockouts have been shown to be genetically identical to parental NOD at unlinked loci and the genetic interval to which recombination has reduced the introgressed B6 or 129 DNA is variable. Frequently, up to 12 generations of backcross have been employed and sometimes microsatellite typing has been used to check for adventitious unlinked non-NOD DNA. In many cases the knockout reduced or eliminated the incidence of diabetes. However, in a minority of cases the knockout (e.g. of PD-1 [22]) has exacerbated disease.
While it is obviously attractive to take advantage of the availability of the rich genetic resource of targeted mutations made on the 129 and B6 backgrounds, the problems of backcrossing require patience, thorough characterization and careful interpretation. In all cases, there will be a significant segment of chromosome surrounding the induced mutation which is derived from the original targeted strain even after 12 generations of backcross; the extent will depend on the distribution of recombination within the relevant genomic region. There may also be other adventitious segments of chromosome inherited from the ES cells or later strains used. Even where the manipulated allele does not lie close to any known diabetes susceptibility/resistance allelic variation, but the targeted allele derives from 129, there may be confounding effects of linked alleles, since most of the known Idd regions have been defined only by variation between NOD and strains other than 129 (mostly B10, B6, NON and NOR). An early example of this problem was provided by the interferon gamma receptor, the knockout of which initially appeared to prevent diabetes in mouse lines backcrossed 10 or 11 generations to NOD. In fact, a single line established at N14 showed that the resistant phenotype was due to a linked gene [23]. No known Idd regions lie on chromosome 10 (where the Ifngr1 gene is found), and the 129 allelic variant responsible for diabetes resistance has not been identified. It should be borne in mind that this problem of confounding effects of linked genes is a general one in mouse knockout studies where the knockout strain has to be backcrossed for functional studies. Studies have shown that the ES-linked region retained is typically 25-40 Mb and that a significant proportion of genes within this congenic segment displays altered transcription levels [24, 25]. Furthermore, sometimes the ES-cell passenger alleles may accelerate diabetes since resistance variants are also present in NOD [26, 27].
The use of knockout alleles originally made in 129 ES cells has, however, been fruitful when combined with transgene rescue. This approach enabled Slattery and colleagues to provide very convincing evidence that beta-2 microglobulin was a diabetes susceptibility locus and probably responsible for the Idd13 resistance region (derived from the NOR strain). While backcross of a beta-2 microglobulin knockout allele from a 129 ES cell targeting event blocked diabetes completely, as might be expected given the effects on development of CD8 T cells, Slattery et al. showed that complementation with a cDNA transgene encoding the B2ma allotype (identical to the NOD form) restored the disease incidence, whereas the B2mb allele (identical to NOR) did not [28]. Both transgenes restored MHC class I protein expression and CD8 development, suggesting a subtle effect on the interaction of MHC class I molecules with peptide and/or their multiple receptors.
4. RNA interference approaches
Kissler has pioneered the use of lentiviral shRNA transgenics as a strategy to investigate the effects of knockdown of specific gene products in the NOD background. This approach has some distinct advantages. First, the transgenic mice can be produced by introducing the foreign DNA directly into the canonical NOD strain, thus avoiding all the problems of inheritance of linked and unlinked alleles from other strains inherent in the backcross approach. Second, in later versions of the lentiviral vector, the expression of the shRNA can be made inducible and reversible by means of an H-tetO promoter. Drawbacks associated with this approach are (a) that knockdown of the target is often incomplete (b) there may be off-target effects of the shRNA and (c) there may be position effects from the inserted DNA. These disadvantages could be mitigated by the use of multiple shRNA sequences and independent transgenic lines with different insertion sites. Kissler and collaborators used this approach to test the hypothesis that the gene Slc11a1 (NRAMP1) is responsible for the susceptibility associated with the Idd5.2 genetic interval, a gene-rich region of 1.48 Mb containing 48 annotated protein encoding genes. Their data strongly supported the notion that the B10 allele at Slc11a1 is responsible for reduction in diabetes incidence in NOD.B10Sn-Idd5.2 congenic mice [29]. This conclusion was assisted by the fact that the B10 allele is believed to encode a null-function protein, despite differing from the functional A/J strain allele by a single amino acid only (position 169), thus rendering the knockdown a near exact phenocopy of the B10 allele. Clearly this will not be the case with most allelic variants.
Perhaps because of this limitation, to date no other NOD susceptibility candidates have been interrogated using RNAi; instead two candidates for human diabetes susceptibility traits have been investigated in the NOD model, variation in soluble CTLA4 expression and an amino acid variant of the phosphatase PTPN22. These knockdowns had opposite effects on disease; partial silencing of sCTLA4 increased disease in NOD.B10Sn-Idd5.1 congenics, though not the parental NOD background [30], and a reduction of 80% in PTPN22 expression reduced diabetes onset in NOD transgenics [31].
5. The development of ES cells from NOD mice
The development of pluripotent mouse cells capable of both genetic modification during in vitro culture and generating novel 'designer' animals has revolutionized mouse genetics. Later, it became apparent that there were potent genetic effects on the ability to isolate such germline-competent embryonic stem (ES) cells. This means that only a few strains of mice could be successfully used to produce such cells by the canonical culture method; most strains, including NOD, are said to be "refractory" with respect to pluripotent ES cell derivation. The first description of the isolation of ES cells from NOD mice, indeed from any spontaneous autoimmune disease model, was provided by Nagafuchi and colleagues in 1999 [32]. They produced 5 ES cell lines by a classical method involving culture of blastocyst-derived cells on primary mouse embryonic fibroblast feeders in the presence of leukemia inhibitory factor (LIF). Only one of these lines retained both ES-like morphology and normal karyotype during serial passage. This ES line was injected into C57BL/6 blastocysts which were reimplanted into pseudopregnant ICR females to produce 22 chimeras ranging from 30% to over 80% ES (NOD) contribution by coat color. However, chimerism, as measured by peripheral blood lymphocyte H-2 allotype, was much lower (<0.2%). Nevertheless, germline transmission was obtained, but only at very low efficiency (1/97 offspring were NOD ES-derived) [32].
Arai and colleagues subsequently made significant improvements to the effectiveness of this first NOD ES line by altering culture conditions. They found that NOD ES cells required a 10-times higher concentration of LIF to prevent differentiation than did 129 (E14.1 and D3) ES lines, and that distinct batches of FCS were optimal for survival and undifferentiated appearance of NOD ES versus 129 ES cells [33]. Most critically, they showed that injection into NOD rather than B6 blastocysts generated chimeras with much higher germline transmission (25-60%). Practically however, making chimeras in NOD blastocysts would be a severe limitation since the screening of chimerism and transmission can only be done by detecting genetic markers introduced into the ES cells. Chen et al. provided a possible solution to this problem by deriving a congenic NOD mouse with an agouti coat. They also isolated their own NOD ES cells. However, while these cells produced chimeras with the NOD.CBALs-Tyr+/Lt blastocysts, they did not transmit through the germline [34]. We are not aware of any targeted mutant NOD mice having been produced by ES cells from either of these studies.
Brook et al. next reported an unsuccessful attempt to derive NOD strain ES cells using microsurgery to explant epiblasts of blastocysts which had been subjected to implantation delay initiated by ovariectomization [35], a technique which they had successfully applied to another refractory strain, CBA/Ca. In the case of NOD mice, few viable blastocysts were recovered; transfer of NOD conceptuses to non-NOD dams produced 9 lines, but all differentiated during passage. They were similarly unsuccessful with NOD congenics which do not develop diabetes. However, when (NOD × 129)F1 embryos were used, the success rate in ES derivation was ~83%, and the derived ES cells made germline competent chimeras at high efficiency [35]. Brook et al. also found that novel congenic strains, made by backcrossing the (NOD ×129)F1 to 129 and subsequent intercross, also made ES cells (at generation N2F4). In one case, these were shown to have good germline competence.
After several years of unsuccessful endeavor, four breakthrough papers describing NOD ES cells appeared in 2009. A critical advance which enabled germline-competent NOD background ES cells to be isolated with high efficiency was made by the discovery in 2008 that ES cells could be maintained in a pluripotent state without LIF or serum by culture in "3i medium" containing 3 small molecule enzyme inhibitors (SU5402 for FGFR, PD184352 for ERK, and CHIR99021 for GSK3) [36]. Ohta et al. used 3i medium to isolate several lines of ES cells from NOD mice [37]. These ES cells were only moderately efficient (2.5-10%) in generating chimeras, but the chimeras themselves gave germline transmission at high frequency (42-100%). Yang et al. used a different inhibitor (SC-1, i.e. pluripotin, which inhibits both ERK1 and Ras-GAP) in combination with LIF to isolate four ES cell lines from NOD-scid mice [38]. It was noticeable that the efficiency with which NOD-scid ES lines produced chimeras was significantly lower than that of other refractory lines (SCID-beige, CD-1). The single successful line gave only 1 chimera from 35 pups born. No data were provided for germline transmission.
Two other independent studies derived more efficient NOD ES cell lines. Hanna et al. used the following inhibitors: (i) Kenpaullone alone, (ii) Kenpaullone in combination with CHIR99021, or (iii) the more typical 2i system (PD184352 + CHIR99021) [39]. Kenpaullone inhibits both GSK3β and CDK1/cyclin B. All three conditions produced NOD ES cells. A single line from each condition was shown to give germline chimeras. Nichols et al. employed yet another 2i system (PD0325901, an MEK inhibitor, and CHIR99021), with LIF to derive NOD ES lines with 53% efficiency. This was considerably higher than that achieved in the conditions used by Hanna et al.; four of these lines were tested by blastocyst injection, and all gave high rates of chimerism (100-60%) and germline transmission (100-75%) [40].
6. Genetic manipulation of NOD ES cells
Obviously, aside from the intrinsic interest in understanding the determinants of embryonic cell pluripotency and differentiation, one of the principal reasons for deriving NOD ES cells is to permit genetic manipulation of NOD mice to further our understanding of the genetics and mechanism of T1D. Thus, it was important to demonstrate that NOD ES cells could be modified, and that mice derived from ES cells would develop diabetes at the expected rate unless intended modifications altered relevant properties. Nichols et al. showed that one NOD ES cell line could be stably transfected with a DsRED expression cassette using PiggyBac transposition, selected by culture in hygromycin, and still give efficient germline competent chimeras [40]. Hanna et al. demonstrated that the Nanog gene could be targeted by homologous recombination in one NOD ES line, but did not report testing chimeric or germline potential after targeting [39]. Nichols et al. also showed that mice derived from unmanipulated NOD ES cells have the same rate of onset and diabetes incidence as 'standard' NOD mice from the same colony [40]. Formally, the incidence cohort of mice, the progeny of NOD-ES→C57BL/6 chimeric males mated with NOD females, was 50% ES-derived. This latter point is of great importance since ES cells in culture may undergo adventitious genetic and epigenetic changes which have the potential to alter diabetes incidence in addition to restricting their germline potential.
There is one very recent publication, by Morgan et al., describing the derivation of genetically targeted NOD mice from pure NOD ES cells [41]. This study shows that the targeted disruption of the H-2DMa gene in NOD mice prevents diabetes onset. Prior to this, there was a report of the derivation of knockin mice from (NOD × 129)F1 ES cells [42]. Kamanaka et al. used homologous targeting to replace exons 1 and 2 (plus intron 1 and part of intron 2) of the Il2 gene with the equivalent B6 genomic DNA. The F1 ES cells were generated by classical means and gave high efficiency chimerism and germline transmission. Ten targeted clones were produced, 3 of which were used to generate subclones in which the inserted floxed neo gene had been removed. All 3 gave rise to germline competent chimeras. However, the use of (NOD × 129)F1 ES cells necessitated two further steps. First, it was essential to determine which parental chromosome in the ES cells had been targeted. This had to be done after breeding the chimeras because of the high degree of local homology between 129 and NOD. A flanking microsatellite showed that two clones had targeted the 129 allele and one the NOD allele. Second, a laborious backcross had to be undertaken to render the genome homozygous for NOD DNA with the chromosome 3 segment containing the targeted Il2 gene. Despite the use of a "speed-backcross" approach, selecting at each generation for the greatest number of NOD alleles at Idd markers, 12 generations were required. The results confirmed that the sequence variation between NOD and B6 at the N-terminus of the IL-2 protein does not alter the incidence of T1D in NOD background mice and cannot be responsible for the B6-Idd3 resistance effect [42], in agreement with the earlier proposal by Yamanouchi et al. [4].
7. The future potential of ES cells and alternative targeting approaches in type 1 diabetes
We will ignore here the potentially important role that both mouse and human ES and iPS cells are likely to play in the development of beta-cell replacement therapies since this topic has been reviewed elsewhere [43]. Instead, we focus on the potential for the exploration of the genetic and mechanistic basis of T1D which is unlocked by targeted germline manipulation. Given the biological resources generated by Hanna and Nichols [39, 40], the NOD mouse field is clearly the most likely to benefit. However, the inhibitor culture approach has also been shown to work for deriving rat ES cells, and it is to be hoped that this will be used on the BB rat model of diabetes [39, 44]. In addition, there has been significant recent progress in genetic modification of rats (and many other diverse organisms) using 'designer nucleases' intended to cleave at unique sites within the genome [45, 46].
Zinc finger nucleases (ZFN) and transcription activator-like effector nucleases (TALENs) represent an alternative approach to gene targeting. This approach is not limited to use in cells with intrinsically high homologous recombination efficiency such as ES cells and can be deployed by direct microinjection of mRNA and/or DNA into zygotes [47-49]. Such designer nucleases have potential for modification of ES cells, including NOD ES cells, and for directly targeting the genome of BB rats and NOD mice. The extent to which this new technology replaces ES cells will depend on the precise type of modification sought and the extent to which problems associated with the use of nuclease, particularly the generation of small deletions or insertions at both the targeted site and at off-target sites elsewhere in the genome can be overcome [50].
We would suggest that exploitation of BB rat germline modification might focus on three avenues where the rat model offers advantages over the mouse:
1. Genetic regions where there may be susceptibility traits common between rats and humans which are absent in mouse (e.g. rat Iddm34 [2])
2. Putative biological pathways which are more experimentally accessible in the larger rodent
3. Studies specifically aimed at understanding those complications of diabetes, e.g. nephropathy, which are better mirrored in the rat model [51].
Furthermore, both NOD mice and BB rats can be used to study other autoimmune pathologies; autoimmune thyroid disease (both models) [52, 53], autoimmune exocrinopathy (AEC; a Sjögren's syndrome-like disease in NOD) [54], and they can be induced to develop autoimmune encephalomyelitis (EAE, NOD) [55] and collagen-induced arthritis (BB rat) [56]. Some genetic susceptibility loci are shared by these diseases. However, other strains may constitute better models, either NOD-related e.g. C57BL/6.NOD-Aec1Aec2 [57] for Sjögren's syndrome, or not directly NOD-related, e.g. Biozzi AB/H for EAE [58]. The 2i culture methodology could be applied to the development of ES cell resources from such autoimmune strains to enable the dissection of these models.
For the NOD mouse, there are three rather obvious directions to be followed (summarized in Figure 1. The first, quickest, and simplest strategic direction will be to use targeted modification of NOD ES cells to confirm or disprove the identity of loci for which we already have good evidence. The genes Il2 (Idd3), Slc11a1 (Idd5.2), Ctla4 (Idd5.1), and Vav3 (Idd18.1) come immediately to mind. In these cases, there is strong evidence, but almost all of it is circumstantial. We are seriously concerned that some authors have confused phenotypic-genotypic correlations, even ones backed up by teleological arguments based on known functions, however plausible, with formal direct tests of hypothetical attribution of diabetes susceptibility genes or even of causative variants. As we discussed above, identification of Idd3 has batted backwards and forwards over time, with protein sequence and transcription levels of IL-2 both favored at different times. Currently the data strongly favor polymorphic variation upstream of the Il2 gene as the causative variants for Idd3. Indeed, del Rio et al. have shown that 6 of the 7 SNP/Indel within the first 1015bp upstream independently influence transcription levels in a reporter assay (albeit one SNP in the opposite direction, NOD>B6) [59]. However, it is quite clear that we cannot say which set of variants between B6 and NOD are responsible for the apparent differences in Il2 transcription rates nor, which is rather more important, can we say for certain that this phenotype is fully responsible for the protection against diabetes offered by B6-Idd3 on the NOD background. We can determine both of these questions with one or more novel targeted NOD strains from NOD ES cells with knock-in mutations. A similar argument can be made for all the Idd for which there are good candidate genes (and in the case of Ctla4 and Slc11a1, candidate variants). Circumstantial evidence, however strong, is not a substitute for proper critical tests.
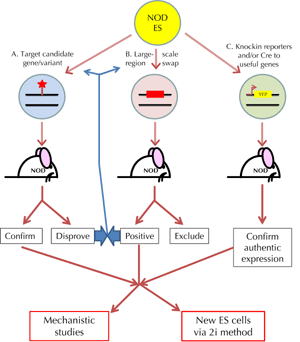 |
 |
Figure 1. Suggested fruitful strategies for the use of NOD strain ES cells. Three general avenues are indicated in the figure. A. NOD ES cells provide a rapid means of confirming or disproving hypothetical candidate genes and causative variants altering diabetes onset by 'knockin' gene targeting. B. Larger scale gene swap technology could be used to refine currently large or unattributed susceptibility regions; the process should be iterative when successful. C. Reporter strains, for example enabling in vivo imaging of beta cells or their progenitors should also be valuable. Cre-expressing strains for lineage-specific modification may also aid mechanistic studies aimed at understanding the biology of diabetes development and generating models for testing interventions. Second generation ES cells derived from gene targeted NOD mice might also be valuable. |
|
The second logical use of NOD ES cells will be to focus down the susceptibility/resistance trait in those Idd regions which are still large and which do not have good candidate genes (see Figure 1). This will require a greater number of targeted ES cells with larger regions 'swapped' for test strain homologous segments and will be more demanding therefore both technically and in resource terms. Perhaps a sensible place to start is to use BAC clones and recombineering technology [60] to replace regions within these large Idd with the relevant resistant strain over those areas which have human orthologous-associated regions (e.g. Idd2). These might then provide both pinpointing information and mechanistic models. This brings us to our third potentially valuable use for NOD ES cells, which is to probe mechanisms of susceptibility/resistance both for mouse-specific-associated genes and to generate models of human disease-associated genes. Even where we have faith that we have identified a genuine disease susceptibility gene, our current understanding of mechanism is weak. If we examine the richness that targeted, especially knockin and conditional knockout, mice have provided in exploring immunity in the broader context, the value that single gene or single causative variant knockin transgenic NOD mice could bring to mechanistic studies of T1D development must be apparent. Furthermore, there is clearly scope for the use of reporter mice on the NOD background, both in mechanistic studies and in setting up models for developing beta-cell replacement therapies. It is worth remembering that the same 2i methodology used to derive NOD ES cells with good efficiency can also be applied to NOD transgenic mice derived from single or multiple targeted ES cells (see Figure 1).
There are clearly some in the diabetes field who question whether any new useful information can be obtained from the NOD mouse model. We believe that there are further valuable lessons to be learned from studying T1D in NOD. Detailed studies aimed at uncovering the exact polymorphisms responsible for genetic susceptibility and resistance to T1D can be addressed through targeted manipulation and in vivo and ex vivo studies of the mouse system. Perhaps more importantly the biological pathways influenced by the susceptibility locus can be investigated and validated in murine models in ways which cannot ethically or affordably be conducted in human subjects. NOD ES cells can not only seal the deal on identification of causative genetic variants, but also permit the development of efficient models for development of candidate therapies.
Disclosure: The authors report no conflict of interests.
Acknowledgments:
We thank Dr Jennifer Nichols for comments on the manuscript. AC acknowledges financial support from the Wellcome Trust.
References
- Leiter EH. Nonobese diabetic mice and the genetics of diabetes susceptibility. Curr Diab Rep 2005. 5:141-148. [DOD] [CrossRef]
- Burren OS, Adlem EC, Achuthan P, Christensen M, Coulson RM, Todd JA. T1DBase: update 2011, organization and presentation of large-scale data sets for type 1 diabetes research. Nucleic Acids Res 2011. 39:D997-D1001. [DOD] [CrossRef]
- Cervino AC, Gosink M, Fallahi M, Pascal B, Mader C, Tsinoremas NF. A comprehensive mouse IBD database for the efficient localization of quantitative trait loci. Mamm Genome 2006. 17:565-574. [DOD] [CrossRef]
- Yamanouchi J, Rainbow D, Serra P, Howlett S, Hunter K, Garner VE, Gonzalez-Munoz A, Clark J, Veijola R, Cubbon R, et al. Interleukin-2 gene variation impairs regulatory T cell function and causes autoimmunity. Nat Genet 2007. 39:329-337. [DOD] [CrossRef]
- Eizirik DL, Sammeth M, Bouckenooghe T, Bottu G, Sisino G, Igoillo-Esteve M, Ortis F, Santin I, Colli ML, Barthson J, et al. The human pancreatic islet transcriptome: expression of candidate genes for type 1 diabetes and the impact of pro-inflammatory cytokines. PLoS Genet 2012. 8:e1002552. [DOD] [CrossRef]
- Acha-Orbea H, McDevitt HO. The first external domain of the nonobese diabetic mouse class II I-Abeta chain is unique. Proc Natl Acad Sci USA 1987. 84:2435-2439. [DOD] [CrossRef]
- Liu GY, Baker D, Fairchild S, Figueroa F, Quartey-Papafio R, Tone M, Healey D, Cooke A, Turk JL, Wraith DC. Complete characterization of the expressed immune response genes in Biozzi AB/H mice: structural and functional identity between AB/H and NOD A region molecules. Immunogenetics 1993. 37:296-300. [DOD]
- Todd JA, Acha-Orbea H, Bell JI, Chao N, Fronek Z, Jacob CO, McDermott M, Sinha AA, Timmerman L, Steinman L, et al. A molecular basis for MHC class II-associated autoimmunity. Science 1988. 240:1003-1009. [DOD] [CrossRef]
- Lund T, O'Reilly L, Hutchings P, Kanagawa O, Simpson E, Gravely R, Chandler P, Dyson J, Picard JK, Edwards A, et al. Prevention of insulin-dependent diabetes mellitus in non-obese diabetic mice by transgenes encoding modified I-A beta-chain or normal I-E alpha-chain. Nature 1990. 345:727-729. [DOD] [CrossRef]
- O'Reilly LA, Healey D, Simpson E, Chandler P, Lund T, Ritter MA, Cooke A. Studies on the thymus of non-obese diabetic (NOD) mice: effect of transgene expression. Immunology 1994. 82:275-286. [DOD]
- Todd JA, Aitman TJ, Cornall RJ, Ghosh S, Hall JR, Hearne CM, Knight AM, Love JM, McAleer MA, Prins JB, et al. Genetic analysis of autoimmune type 1 diabetes mellitus in mice. Nature 1991. 351:542-547. [DOD] [CrossRef]
- Prins JB, Todd JA, Rodrigues NR, Ghosh S, Hogarth PM, Wicker LS, Gaffney E, Podolin PL, Fischer PA, Sirotina A, Peterson LB. Linkage on chromosome 3 of autoimmune diabetes and defective Fc receptor for IgG in NOD mice. Science 1993. 260:695-698. [DOD] [CrossRef]
- Podolin PL, Denny P, Lord CJ, Hill NJ, Todd JA, Peterson LB, Wicker LS, Lyons PA. Congenic mapping of the insulin-dependent diabetes (Idd) gene, Idd10, localizes two genes mediating the Idd10 effect and eliminates the candidate Fcgr1. J Immunol 1997. 159:1835-1843. [DOD]
- Lyons PA, Armitage N, Argentina F, Denny P, Hill NJ, Lord CJ, Wilusz MB, Peterson LB, Wicker LS, Todd JA. Congenic mapping of the type 1 diabetes locus, Idd3, to a 780-kb region of mouse chromosome 3: identification of a candidate segment of ancestral DNA by haplotype mapping. Genome Res 2000. 10:446-453. [DOD] [CrossRef]
- Spolski R, Kashyap M, Robinson C, Yu Z, Leonard WJ. IL-21 signaling is critical for the development of type I diabetes in the NOD mouse. Proc Natl Acad Sci USA 2008. 105:14028-14033. [DOD] [CrossRef]
- Sutherland AP, Van Belle T, Wurster AL, Suto A, Michaud M, Zhang D, Grusby MJ, von Herrath M. Interleukin-21 is required for the development of type 1 diabetes in NOD mice. Diabetes 2009. 58:1144-1155. [DOD] [CrossRef]
- McGuire HM, Walters S, Vogelzang A, Lee CM, Webster KE, Sprent J, Christ D, Grey S, King C. Interleukin-21 is critically required in autoimmune and allogeneic responses to islet tissue in murine models. Diabetes 2011. 60:867-875. [DOD] [CrossRef]
- Van Belle TL, Nierkens S, Arens R, von Herrath MG. Interleukin-21 receptor-mediated signals control autoreactive T cell infiltration in pancreatic islets. Immunity 2012. 36:1060-1072. [DOD] [CrossRef]
- Schorle H, Holtschke T, Hünig T, Schimpl A, Horak I. Development and function of T cells in mice rendered interleukin-2 deficient by gene targeting. Nature 1991. 352:621-624. [DOD] [CrossRef]
- Thomas KR, Capecchi MR. Site-directed mutagenesis by gene-targeting in mouse embryo-derived stem cells. Cell 1987. 51:505-512. [DOD] [CrossRef]
- Malissen M, Gillet A, Ardouin L, Bouvier G, Trucy J, Ferrier P, Vivier E, Malissen B. Altered T cell development in mice with a targeted mutation of the CD3-epsilon gene. EMBO J 1995. 14:4641-4653. [DOD]
- Wang J, Yoshida T, Nakaki F, Hiai H, Okazaki T, Honjo T. Establishment of NOD-Pdcd1-/- mice as an efficient animal model of type I diabetes. Proc Natl Acad Sci USA 2005. 102:11823-11828. [DOD] [CrossRef]
- Kanagawa O, Xu G, Tevaarwerk A, Vaupel BA. Protection of nonobese diabetic mice from diabetes by gene(s) closely linked to IFN-g receptor loci. J Immunol 2000. 164:3919-3923. [DOD]
- Schalkwyk LC, Fernandes C, Nash MW, Kurrikoff K, Vasar E, Koks S. Interpretation of knockout experiments: the congenic footprint. Genes Brain Behav 2007. 6:299-303. [DOD] [CrossRef]
- Valor LM, Grant SG. Clustered gene expression changes flank targeted gene loci in knockout mice. PLoS One 2007. 2:e1303. [DOD] [CrossRef]
- McAleer MA, Reifsnyder P, Palmer SM, Prochazka M, Love JM, Copeman JB, Powell EE, Rodrigues NR, Prins JB, Serreze DV, et al. Crosses of NOD mice with the related NON strain. A polygenic model for IDDM. Diabetes 1995. 44:1186-1195. [DOD] [CrossRef]
- Gonzalez A, Katz JD, Mattei MG, Kikutani H, Benoist C, Mathis D. Genetic control of diabetes progression. Immunity 1997. 7:873-883. [DOD] [CrossRef]
- Hamilton-Williams EH, Serreze DV, Charlton B, Johnson EA, Marron MP, Mullbacher A, Slattery RM. Transgenic rescue implicates beta2-microglobulin as a diabetes susceptibility gene in nonobese diabetic (NOD) mice. Proc Natl Acad Sci USA 2001. 98:11533-11538. [DOD] [CrossRef]
- Kissler S, Stern P, Takahashi K, Hunter K, Peterson LB, Wicker LS. In vivo RNA interference demonstrates a role for Nramp1 in modifying susceptibility to type 1 diabetes. Nat Genet 2006. 38:479-483. [DOD] [CrossRef]
- Gerold KD, Zheng P, Rainbow DB, Zernecke A, Wicker LS, Kissler S. The soluble CTLA-4 splice variant protects from type 1 diabetes and potentiates regulatory T-cell function. Diabetes 2011. 60:1955-1963. [DOD] [CrossRef]
- Zheng P, Kissler S. PTPN22 Silencing in the NOD Model Indicates the Type 1 Diabetes-Associated Allele Is Not a Loss-of-Function Variant. Diabetes 2012. In press. [DOD]
- Nagafuchi S, Katsuta H, Kogawa K, Akashi T, Kondo S, Sakai Y, Tsukiyama T, Kitamura D, Niho Y, Watanabe T. Establishment of an embryonic stem (ES) cell line derived from a non-obese diabetic (NOD) mouse: in vivo differentiation into lymphocytes and potential for germ line transmission. FEBS Lett 1999. 455:101-104. [DOD] [CrossRef]
- Arai S, Minjares C, Nagafuchi S, Miyazaki T. Improved experimental procedures for achieving efficient germ line transmission of nonobese diabetic (NOD)-derived embryonic stem cells. Exp Diabesity Res 2004. 5:219-226. [DOD] [CrossRef]
- Chen J, Reifsnyder PC, Scheuplein F, Schott WH, Mileikovsky M, Soodeen-Karamath S, Nagy A, Dosch MH, Ellis J, Koch-Nolte F, Leiter EH. "Agouti NOD": identification of a CBA-derived Idd locus on Chromosome 7 and its use for chimera production with NOD embryonic stem cells. Mamm Genome 2005. 16:775-783. [DOD] [CrossRef]
- Brook FA, Evans EP, Lord CJ, Lyons PA, Rainbow DB, Howlett SK, Wicker LS, Todd JA, Gardner RL. The derivation of highly germline-competent embryonic stem cells containing NOD-derived genome. Diabetes 2003. 52:205-208. [DOD] [CrossRef]
- Ying QL, Wray J, Nichols J, Batlle-Morera L, Doble B, Woodgett J, Cohen P, Smith A. The ground state of embryonic stem cell self-renewal. Nature 2008. 453:519-523. [DOD] [CrossRef]
- Ohta H, Ohinata Y, Ikawa M, Morioka Y, Sakaide Y, Saitou M, Kanagawa O, Wakayama T. Male germline and embryonic stem cell lines from NOD mice: efficient derivation of GS cells from a nonpermissive strain for ES cell derivation. Biol Reprod 2009. 81:1147-1153. [DOD] [CrossRef]
- Yang W, Wei W, Shi C, Zhu J, Ying W, Shen Y, Ye X, Fang L, Duo S, Che J, et al. Pluripotin combined with leukemia inhibitory factor greatly promotes the derivation of embryonic stem cell lines from refractory strains. Stem Cells 2009. 27:383-389. [DOD] [CrossRef]
- Hanna J, Markoulaki S, Mitalipova M, Cheng AW, Cassady JP, Staerk J, Carey BW, Lengner CJ, Foreman R, Love J, et al. Metastable pluripotent states in NOD-mouse-derived ESCs. Cell Stem Cell 2009. 4:513-524. [DOD] [CrossRef]
- Nichols J, Jones K, Phillips JM, Newland SA, Roode M, Mansfield W, Smith A, Cooke A. Validated germline-competent embryonic stem cell lines from nonobese diabetic mice. Nat Med 2009. 15:814-818. [DOD] [CrossRef]
- Morgan MA, Muller PS, Mould A, Newland SA, Nichols J, Robertson EJ, Cooke A, Bikoff EK. The Nonconventional MHC class II molecule DM governs diabetes susceptibility in NOD mice. PLoS One 2013. 8:e56738. [DOD] [CrossRef]
- Kamanaka M, Rainbow D, Schuster-Gossler K, Eynon EE, Chervonsky AV, Wicker LS, Flavell RA. Amino acid polymorphisms altering the glycosylation of IL-2 do not protect from type 1 diabetes in the NOD mouse. Proc Natl Acad Sci USA 2009. 106:11236-11240. [DOD] [CrossRef]
- Liew CG, Andrews PW. Stem cell therapy to treat diabetes mellitus.. Rev Diabet Stud 2008. 5(4):203-219. [DOD] [CrossRef]
- Wallis RH, Wang K, Marandi L, Hsieh E, Ning T, Chao GY, Sarmiento J, Paterson AD, Poussier P. Type 1 diabetes in the BB rat: a polygenic disease. Diabetes 2009. 58:1007-1017. [DOD] [CrossRef]
- Geurts AM, Cost GJ, Freyvert Y, Zeitler B, Miller JC, Choi VM, Jenkins SS, Wood A, Cui X, Meng X, et al. Knockout rats via embryo microinjection of zinc-finger nucleases. Science 2009. 325:433. [DOD] [CrossRef]
- Mashimo T, Takizawa A, Voigt B, Yoshimi K, Hiai H, Kuramoto T, Serikawa T. Generation of knockout rats with X-linked severe combined immunodeficiency (X-SCID) using zinc-finger nucleases. PLoS One 2010. 5:e8870. [DOD] [CrossRef]
- Cui X, Ji D, Fisher DA, Wu Y, Briner DM, Weinstein EJ. Targeted integration in rat and mouse embryos with zinc-finger nucleases. Nat Biotechnol 2011. 29:64-67. [DOD] [CrossRef]
- Tesson L, Usal C, Ménoret S, Leung E, Niles BJ, Remy S, Santiago Y, Vincent AI, Meng X, Zhang L, et al. Knockout rats generated by embryo microinjection of TALENs. Nat Biotechnol 2011. 29:695-696. [DOD] [CrossRef]
- Meyer M, Ortiz O, Hrabe de Angelis M, Wurst W, Kühn R. Modeling disease mutations by gene targeting in one-cell mouse embryos. Proc Natl Acad Sci USA 2012. 109:9354-9359. [DOD] [CrossRef]
- Wang J, Friedman G, Doyon Y, Wang NS, Li CJ, Miller JC, Hua KL, Yan JJ, Babiarz JE, Gregory PD, Holmes MC. Targeted gene addition to a predetermined site in the human genome using a ZFN-based nicking enzyme. Genome Res 2012. 22:1316-1326. [DOD] [CrossRef]
- Cohen AJ, McGill PD, Rossetti RG, Guberski DL, Like AA. Glomerulopathy in spontaneously diabetic rat. Impact of glycemic control. Diabetes 1987. 36:944-951. [DOD] [CrossRef]
- Many MC, Maniratunga S, Denef JF. The non-obese diabetic (NOD) mouse: an animal model for autoimmune thyroiditis. Exp Clin Endocrinol Diabetes 1996. 104(Suppl 3):17-20. [DOD] [CrossRef]
- Allen EM, Appel MC, Braverman LE. The effect of iodide ingestion on the development of spontaneous lymphocytic thyroiditis in the diabetes-prone BB/W rat. Endocrinology 1986. 118:1977-1981. [DOD] [CrossRef]
- Thompson C, Jacobsen H, Pomeranz Krummel D, Nagai K, Cooke A. Non-depleting anti-CD4 antibody not only prevents onset but resolves sialadenitis in NOD mice. Autoimmunity 2004. 37:549-554. [DOD] [CrossRef]
- Bernard CC, Johns TG, Slavin A, Ichikawa M, Ewing C, Liu J, Bettadapura J. Myelin oligodendrocyte glycoprotein: a novel candidate autoantigen in multiple sclerosis. J Mol Med (Berl) 1997. 75:77-88. [DOD] [CrossRef]
- Smith RJ, Sly LM. Type II collagen-induced arthritis in the diabetic-resistant biobreeding rat: inflammatory and histopathological features of joint pathology and effects of antiinflammatory and antirheumatic drugs on this chronic arthritic process. J Pharmacol Exp Ther 1996. 277:1801-1813. [DOD]
- Cha S, Nagashima H, Brown VB, Peck AB, Humphreys-Beher MG. Two NOD Idd-associated intervals contribute synergistically to the development of autoimmune exocrinopathy (Sjögren's syndrome) on a healthy murine background. Arthritis Rheum 2002. 46:1390-1398. [DOD] [CrossRef]
- Baker D, Rosenwasser OA, O'Neill JK, Turk JL. Genetic analysis of experimental allergic encephalomyelitis in mice. J Immunol 1995. 155:4046-4051. [DOD]
- del Rio R, Noubade R, Subramanian M, Saligrama M, Diehl S, Rincon M, Teuscher C. SNPs upstream of the minimal promoter control IL-2 expression and are candidates for the autoimmune disease-susceptibility locus Aod2/Idd3/Eae3. Genes and Immunity 2008. 9:115-121. [DOD] [CrossRef]
- Testa G, Zhang Y, Vintersten K, Benes V, Pijnappel WW, Chambers I, Smith AJ, Smith AG, Stewart AF. Engineering the mouse genome with bacterial artificial chromosomes to create multipurpose alleles. Nat Biotechnol 2003. 21:443-447. [DOD] [CrossRef]
This article has been cited by other articles:
|


)


