Review
| Rev Diabet Stud,
2008,
5(4):203-219 |
DOI 10.1900/RDS.2008.5.203 |
Stem Cell Therapy to Treat Diabetes Mellitus
Chee Gee Liew1, Peter W. Andrews2
1Sue and Bill Gross Stem Cell Research Center, 101 Theory, University of California, Irvine, California 92617, USA
2Centre for Stem Cell Biology, Department of Biomedical Science, The University of Sheffield, Western Bank, Sheffield S10 2TN, UK
Address correspondence to: Chee Gee Liew, e-mail: cliew@uci.edu
Manuscript submitted November 10, 2008; resubmitted January 26, 2009; accepted February 9, 2009.
Keywords: type 1 diabetes, stem cell, beta-cell, transplantation, Pdx1, Sox9, NeuroD1, ngn3
Abstract
Transplantation of pancreatic islets offers a direct treatment for type 1 diabetes and in some cases, insulin-dependent type 2 diabetes. However, its widespread use is hampered by a shortage of donor organs. Many extant studies have focused on deriving β-cell progenitors from pancreas and pluripotent stem cells. Efforts to generate β-cells in vitro will help elucidate the mechanisms of β-cell formation and thus provide a versatile in vivo system to evaluate the therapeutic potential of these cells to treat diabetes. Various successful experiments using β-cells in animal models have generated extensive interest in using human embryonic stem cells to restore normoglycemia in diabetic patients. While new techniques are continually unveiled, the success of β-cell generation rests upon successful manipulation of culture conditions and the induction of key regulatory genes implicated in pancreas development. In this review, we compare successfully conducted protocols, highlight essential steps and identify some of the remarkable shortfalls common to these methods. In addition, we discuss recent advancements in the derivation of patient-specific pluripotent stem cells that may facilitate the use of autologous β-cells in stem cell therapy.
Diabetes and islet cell transplantation
Diabetes mellitus is a chronic metabolic disorder manifested by hyperglycemia due to a deficiency of insulin production by pancreatic β-cells. This can be a direct consequence of autoimmune destruction of β-cells, as seen in type 1 diabetes (T1D) [1-3]. Other types of diabetes, collectively known as type 2 diabetes (T2D), occur because of a combination of reduced insulin sensitivity (non-insulin dependent) and impaired β-cells function [4]. Diabetes can be inherited, such as that seen in maturity onset diabetes of the young adult (MODY), or caused by mutations in an autosomal dominant gene resulting in the disruption of insulin production. Gestational diabetes is another less known form of diabetes, but it imposes an increasingly prevalent risk factor for the development of T2D after pregnancy [5].
Exogenous insulin supply is required to maintain normoglycemia in many diabetic patients. A promising way of treatment is using β-cell replacement therapy. For successful islet transplantation a sufficient amount of β-cells needs to be included in the transplant to control the blood glucose level without repeated insulin injections. In 2000, Shapiro and colleagues described the successful cure of T1D in a small number of patients using a procedure known as the 'Edmonton Protocol' [6]. The authors achieved independence of insulin injections in seven T1D patients by transplanting a large number of islet cells (obtained from two donor pancreases) combined with the use of glucocorticoid-free immunosuppressive regimen. Notably, both host-versus-graft and autoimmune reactions were avoided. However, despite short term success, long term insulin independence is usually not sustainable. Their international clinical trial showed that only 13% of the patients (5 out of 36 subjects with T1D) maintained insulin independence at 2 years, and 28% had complete graft loss 1 year after the final transplantation [7].
One explanation for the poor long-term outcome is recurrent immune destruction of the transplanted islets despite immunosuppression. Indeed, late graft loss is commonly observed although graft function measured by C-peptide immunoreacticity was retained. This phenomenon of ‘islet graft exhaustion’ could be attributed to the toxicity of immunosupression, autorejection or recurrence of autoimmunity [8]. Long term failure of the early transplantation therapies has prompted the search for more defined sources of β-cells. Intensive research is being conducted to look for alternative sources of β-cells because of the shortage of cadaver donors. An alternative resource for transplantable β-cells is the stem cell. This article reviews the current knowledge on the role of stem cells in pancreatic development and as a source for β-cell transplantation. In addition, it discusses recent advancements in the derivation of patient-specific pluripotent stem cells that may facilitate the use of autologous β-cells in stem cell therapy.
Formation of new β-cells: knowledge from developmental biology of the pancreas
Stem cells are cells that are able to proliferate while maintaining an undifferentiated status (self-renewal) and retaining a capacity to differentiate into specialized cell types under appropriate conditions. The sources of such stem cells for the purpose of generating β-cells were firstly identified through several studies on adult and fetal pancreas. It is generally assumed that islet neogenesis or replication occurs throughout life with a gradual decrease in capacity in elder humans. It is also assumed that the generation of β-cells can take place by two pathways: firstly via replication of already existing β-cells and secondly via neogenesis (differentiation) from pancreatic endocrine or ductal progenitors [9, 10]. During fetal life, the majority of new β-cells develop from precursors, but newly developed β-cells also proliferate. In the adult pancreas, the rate of both β-cell neogenesis and replication is more limited than in newborn, but it is thought to take place [11-13]. However, a study by Dor et al. (2004) has challenged the concept of neogenesis, because the authors did not observe new β-cells formed from non-insulin-producing stem cells during adult life [14].
Early pattern of differentiation in the mouse and human pancreas
During early embryogenesis, the epiblast and primitive endoderm arise from the inner cell mass (ICM) of the blastocyst, from which pluripotent embryonic stem (ES) cells are derived. The primitive endoderm gives rise to extraembryonic tissue, whereas the epiblast differentiates to form the three primary germ layers during gastrulation. Cells from the epiblast are recruited into the primitive streak and migrate out to form the mesoderm and definitive endoderm (DE), from which the pancreas develops. Nodal and activin, members of transforming growth factor beta (TGF-β) superfamily, are essential for the initial endoderm specification, where they regulate the activation of many key regulatory genes, such as foxa2, sox17 and mixl1 in a strict temporal sequence. The primitive gut tube then arises from the DE, which later forms foregut, midgut and hindgut [15]. Signals from the notochord and mesenchyme specify the pancreatic domain at the endodermal region. The pancreas develops from the posterior foregut and the subsequent formation of the pancreatic anlagen relies on retinoid signaling and on inhibition of Indian and Sonic hedgehog (Shh) signaling. The developing pancreas is composed of pancreatic and duodenal homeobox 1 (Pdx1)-expressing epithelial precursor cells that give rise to the three differentiated compartments of endocrine, exocrine and ductal cells found in the adult pancreas. The endocrine cell mass aggregates in interstitial clusters adjacent to the ductal epithelial cells, to form the islets of Langerhans [16].
In mice, progression from foregut endoderm to insulin-producing cells is rapid (between E9.5 and E10). Nonetheless, in humans, although pancreatic differentiation from foregut endoderm is initiated at an equivalent time in human (26 days post conception, dpc), significant insulin expression is delayed. This observation supports the notion that embryological stages in the mouse and the human are not as closely equivalent as previously assumed [17]. In humans, by 35 dpc, the ventral pancreatic bud begins to migrate backwards and comes into contact and eventually fuses with the dorsal pancreatic bud during 6 weeks post conception (wpc) [18]. Both insulin and glucagon expression could only be detected at 8 wpc [19, 20], two weeks later than expected compared to the mouse [21].
Insulin is the most abundant hormone detected during the first trimester in human pancreas [22]. At 10 wpc, all four islet hormones can be detected. Islets of Langerhans are formed at 12-13 wpc. At this stage, islets are comprehensively vascularized and contain cells independently immunoreactive for insulin, glucagon, somatostatin and pancreatic polypeptide (PP). This is accompanied by the expression of prohormone convertase 1/3 (PC1/3), islet amyloid polypeptide (IAPP) and β-cell specific glucose transporter-2 (GLUT2) [22]. The expression of these markers implies that fetal β-cells may be capable of processing and secreting insulin. However, the human fetal pancreas is not glucose-responsive, despite being able to secrete insulin upon stimulation with β-cell secretagogues [23]. These studies notwithstanding, the mechanisms that control endocrine cell formation from human neonatal pancreas are poorly understood because of the apparent scarcity of material and difficulties in obtaining human fetuses.
Pancreas and islet-related transcription factors
In mice, embryonic pancreatic epithelial cells express nuclear Pdx1 and cytoplasmic cytokeratin-19 (CK19) [22]. Pdx1 plays an important role in the transactivation of the insulin gene and so is required to maintain normal β-cell homeostasis [21]. Another more recently described pancreas-related transcription factor, Sox9, is predominantly expressed throughout the early developing pancreas (prior to 14 weeks of gestation). In contrast to Pdx1, the expression of Sox9 is down-regulated once endocrine cells are developed and is later restricted to ductal cells. A study using sox9 heterozygous mouse mutants suggests that the role of sox9 is as a determinant of multipotent pancreatic endocrine cells in the pancreas [24].
Pancreatic endocrine cell fate specification is also ensured by a lateral inhibition process mediated by Notch signaling pathways. Genetic studies that involved ectopic expression of neurogenin3 (ngn3) and intracellular Notch in early pancreas progenitors collectively confirm the function of ngn3 in controlling endocrine cell fate. Mice lacking ngn3 function fail to generate pancreatic endocrine cells and die postnatally from diabetes [25]. Similarly, neuroD1 knock-out mice fail to develop islets and develop severe diabetic ketoacidosis and perinatal death [26]. It has been shown that maturity onset diabetes of the young type 6 (MODY-6) in humans is also associated with mutations in NEUROD1 and that the abnormality of islet morphogenesis is due in part to inadequate expression of the INSULIN gene [27].
Islet1 expression in pancreatic endodermal cells is required for the formation of dorsal mesenchyme and generation of all endocrine islet cells. A number of genes control the differentiation of specific pancreatic endocrine cell subsets. Pax4 is required for the initial commitment of early endocrine precursors to become β- and δ-cells, while pax6 is required for the early differentiation of α-cells [28-33]. Fully differentiated β-cells first appear around E13 at the start of a massive wave of β-cell differentiation, which is known as “secondary transition” [34]. Nkx6.1 expression is required by this second phase of β-cell neogenesis in the developing pancreas [35, 36]. A recently described transcription factor, MafA is induced at the final stage of β-cell differentiation and functions as a potent activator of insulin gene transcription [37].
Approximately 90% of β-cells and 15% of δ-cells in adult islets express Pdx1. Pdx1 regulates the expression of β-cell-specific genes such as INSULIN, IAPP (islet amyloid polypeptide), β-cell-specific glucose transporters glucokinase (GCK) and GLUT2. Hence, the expression of Pdx1, Gck and GLUT2 can be used as a key indicator of β-cell functionality [38]. The dual action of Pdx1, as a pancreas commitment factor during embryogenesis and as a regulator of islet cell physiology in mature islet cells, underscores the unique role of PDX1 in maintaining the function of human pancreatic endocrine cells [39]. Apart from PDX1, mutations of which have been linked to MODY-4 [40, 41], several other genes have also been shown to be required in humans and in mice to assure β-cell functionality. These include the other five MODY genes: GCK (MODY-2), hepatocyte nuclear factor 1a (HNF1A; MODY-3), HNF1B (MODY 5), HNF4A (MODY-1) and NEUROD1 (MODY-6) [27, 42].
Properties of a mature β-cell and insulin biosynthesis
The gold standard for defining β-cell function is glucose responsiveness. A functional β-cell exhibits an acute three-fold stimulatory insulin release in response to glucose. Zinc is required for packaging insulin, an integral part of insulin crystals for 2-Zn-insulin hexamer, as well as free ionized zinc in the extragranular space that acts as a reservoir for granular zinc pools [43-46]. The ability to regulate glucose uptake by the islet-specific glucose transporter GLUT2 is the first step necessary for the activation of the regulatory region of the Insulin gene to glucose [47]. In the absence of GLUT2, the endocrine pancreas shows the loss of first phase glucose-stimulated insulin secretion and an inverse α- to β-cell ratio [48]. Glucose signaling to secretion and insulin biosynthesis are also impaired [49]. The synthesis of the glucose-phosphorylating enzyme glucokinase is a late event in β-cell maturation [50]. Patients with mutations of the GCK have mild fasting hyperglycemia throughout life [42].
Insulin mRNA is translated as a single chain precursor called preproinsulin, and the removal of its signal peptide during insertion into the endoplasmic reticulum generates proinsulin. Proinsulin consists of three domains: an amino-terminal B chain (30 amino acids), a carboxy-terminal A chain (21 amino acids) and a connecting peptide in the middle known as the C-peptide. Within the endoplasmic reticulum, proinsulin is exposed to several specific endopeptidases that excise the C-peptide, thereby generating the mature form of insulin. Insulin and free C-peptide are packaged in the Golgi into secretory granules, which accumulate in the cytoplasm. In pancreatic β-cells, C-peptide and mature insulin are present in equimolar ratios and co-localize in the secretory granules [51].
Sources of renewable β-cells
Pancreatic stem cells in pancreas ductal cells
Several studies have revealed the possibility that endocrine precursors lie within pancreatic ducts. In the adult pancreas, endocrine cell formation from ductal epithelial cells has been observed both in experimental models of pancreas injury [52] and in various clinical pathologies [53]. Islet neogenesis and replication may provide the source of pancreatic stem cells from which normal renewal of islets occurs throughout life [54].
Ramiya et al. (2000) generated pancreatic islets from adult murine ductal cells in vitro [55]. The pancreatic stem cells generated insulin-producing stem cells (IPSC) in a monolayer, from which islet progenitor cells (IPC) budded. The pancreatic islet mass significantly increased by growing IPC in culture; many of the cells expressed both glucagon and insulin, a phenotype reported for immature islet cells on their path to end-stage differentiation [56]. These IPSC-derived islets secreted insulin in response to glucose challenge, and upon addition of nicotinamide they further matured and differentiated into fully functional islets. After transplantation, the mice were able to regulate the levels of glucose in their blood within a week, and survive without further need for insulin.
Bonner-Weir et al. (2000) demonstrated that human pancreas duct tissue can also be expanded in vitro and then be directed to differentiate into glucose-responsive islet buds after being overlaid with matrigel [57]. The cells were grown in serum-free media with glucose, insulin, transferrin and selenium (ITS) supplements, nicotinamide and keratinocyte growth factor (KGF). The epithelial cells formed three-dimensional cystic structures, characteristic of cultivated human islet buds. These cultivated human islet buds consisted of both CK19-positive ductal cells and hormone-positive islet cells. There was a significant increase in DNA and insulin content over 3-4 weeks of culture, suggesting that these human islet buds were immature and still in the process of differentiation.
Hui et al. (2001) demonstrated that overexpression of Pdx1 and treatment with glucagon-like peptide 1 (GLP-1) induce the differentiation of rat and human pancreas ducts into insulin-secreting cells [50]. GLP-1 is capable of restoring normal glucose tolerance in aging mice and increasing islet mass in adult animals previously subjected to subtotal pancreatectomy [9]. Additionally, endogenously pdx1-positive rat (ARIP) and stably pdx1-transfected human (PANC-1) cell lines attained a β-cell phenotype upon GLP-1 treatment. This study provides a significant basis for determining the minimum biological requirements for a non-β-cell to become a β-cell, whilst supporting the previous finding that pancreatic ductal cell lines are capable of giving rise to pancreatic endocrine cells.
Guided by the expression of ngn3, β-cell precursors were identified in an injury model of adult pancreas [58]. Following partial duct ligation, cells located in the ductal lining were found to reactivate ngn3. These Ngn3-positive cells express ductal cytokeratins, but did not stain for insulin, suggesting they are of islet progenitor phenotype. However, they developed into cells that subsequently proliferated, both in situ and in vitro, and were found to also express Pdx1. Ngn3-positive cells were then isolated and injected into ex vivo embryonic mouse pancreas, and were found to autonomously (not by fusion) increase glucose-responsive β-cell mass in the explants.
Dor et al. (2004) have challenged the concept of neogenesis despite the aforementioned evidence of the existence of β-cell precursors [14]. To prove this, they labeled β-cells in transgenic mice with insulin promoter driving an inducible Cre/lox system. Following tamoxifen induction, a human placental alkaline phosphatase (HPAP) reporter gene was expressed ('pulse'). Thus, during turnover ('chase'), β-cells could be identified with HPAP dye. Surprisingly, all islets analyzed in adult pancreas in the ‘pulse and chase’ experiments contained numerous HPAP-immunoreactive β-cells. Nevertheless, no insulin-immunoreactive and HPAP-negative cells were observed. Notably, partial pancreatectomy resulted in the same observation but with increased bromodeoxyuridine (BrdU) incorporation, indicating that these β-cells retained their full proliferative capacity following injury. Thus, their observation indicates that terminally differentiated β-cells are capable of self-renewal and differentiation into new β-cells. To date, this study remains controversial because it does not prove the absence of stem cells during neonatal life or after pancreas injury, and the possible transcriptional activity of insulin promoter in such overlooked stem cells.
Pancreatic stem cells in islets of Langerhans
The endocrine cells of the adult rat pancreatic islets of Langerhans, including β-cells, turn over every 40-50 days by a process of apoptosis, and are replaced by neogenesis from progenitor epithelial cells located in the pancreatic ducts [55, 57]. However, the administration of glucose or GLP-1 to rats for 48 hours resulted in a doubling of islet cell mass, suggesting that islet progenitor cells were able to proliferate and that the precursors may reside within the islets themselves [59].
In another study, rat and human pancreatic islets were isolated post-mortem and cultivated in medium supplemented by growth factors. Focal regions of nestin-positive cells were identified in large, small, and centrolobular ducts of the rat pancreas. These nestin-expressing cells were not ductal epithelial cells as they were CK19-negative. When these cells were grown in medium containing basic fibroblast growth factor (bFGF) and epidermal growth factor (EGF), they were able to proliferate. When the medium was switched to low glucose medium containing hepatocyte growth factor (HGF), betacellulin, activin-A, exendin-4 or nicotinamide, they displayed a ductal/endocrine phenotype with expression of CK19, neural-specific cell adhesion molecule (NCAM), insulin, glucagon, glut2 and pdx1, besides the expression of liver and exocrine pancreas markers. These findings provide evidence that pancreatic islets themselves, apart from the ducts contain multipotential progenitor cells that are able to differentiate to β-cells.
However, controversy exists regarding the significance of nestin-expressing cells in islet neogenesis. Selander and Edlund did not find nestin expression in the pancreatic ductal epithelium [60], where the potential progenitors reside. Instead, nestin was detected in mesenchymal cells, called pancreatic stellate cells [61, 62] and was not detected during pancreas development [21]. Nonetheless, many groups have tested nestin selection protocols to derive β-like cells from mouse embryonic stem cells (MESC) later.
Pancreas and β-cell precursors from exocrine tissue
Human cells derived ex vivo from pancreatic exocrine tissue isolated from healthy donors de-differentiated into a ductal phenotype (CK19- and CK7-positive) after adherence to culture surfaces [63]. After 2 days in culture, these cells reactivated PDX1 expression, suggesting their pancreatic precursor potential; PDX1 re-expression in ductal cells has been considered to be a prerequisite for their differentiation. This phenomenon of 'transdifferentiation' is questionable, since these in vitro studies are subject to ductal cell contamination [64]. However, a recent report has demonstrated that pancreatic exocrine cells can rapidly adopt a β-cell phenotype when the genome of these cells was altered and reprogrammed [65].
Zhou et al. (2008) developed a strategy to re-express multiple embryonic genes using adenoviruses in pancreatic exocrine cells of adult mice [65]. Nine genes that exhibit β-cell phenotypes when mutated were selected for initial reprogramming experiments. Of these, the combination of Ngn3, Pdx1 and MafA transcription factors could efficiently reprogram 20% of differentiated pancreatic exocrine cells into insulin-positive β-cells in vivo. Although emergent extra-islet insulin-positive cells did not appear to self-cluster as islets, angiogenesis was remarkably increased. Importantly, exocrine cells that adopted a β-cell phenotype improved hyperglycemia in STZ-induced diabetic mice. Taken together, those studies suggest that fully differentiated or even somatic cells could serve as an important source of tissue for generating functional insulin-producing cells in diabetic patients. Zhou’s study was largely based on earlier studies that have provided the evidence that these lineage-committed cells can further be reprogrammed back to an ES cell fate, a characteristic termed induced pluripotency, or more commonly referred to as reprogramming.
β-cells from other adult tissues
The ability of adult stem cells to produce differentiated cells from embryologically unrelated tissues is an example of metaplasia and shows that embryological commitments can be changed or reversed under certain circumstances [66]. For example, both rodent and human bone-marrow stromal cells have been demonstrated to differentiate into other mesodermally derived tissues such as cardiac muscle cells [67], liver or neurons [68, 69], which are ectodermal in origin.
Although no significant in vivo differentiation of bone marrow into β-cells was observed in a study of adult mice [70], transdifferentiation from liver to pancreas has been seen. Ferber et al. (2000) demonstrated that transdifferentiation of liver to pancreas could be induced by ectopic expression of pdx1; transient expression of pdx1 in mouse liver cells in vivo induced expression of the otherwise silent endogenous insulin gene [71]. A follow-up study indicated that insulin- as well as glucagon-producing cells were found mainly located in the proximity of central veins [72]. This indicates that pdx1 plays an important instructive role in pancreas differentiation, not only from primitive gut endoderm but also to reprogram extra-pancreatic tissue towards a β-cell phenotype [72]. It was also sufficient to direct the production and secretion of mature, biologically active insulin from a restricted population of cells in liver in vivo [71]. Insulin secreted from the liver of pdx1-transfected mice ameliorated streptozotocin (STZ)-induced diabetes.
β-cell precursors from fetal pancreas
Islet-like cell clusters (ICC) cultured from human fetal pancreatic tissue (18 to 24 weeks gestation) were able to mature functionally and morphologically when grafted to kidney or pancreas in normoglycemic mice [73]. Additionally, exendin-4, used to treat those human fetal pancreas-derived ICC in vitro, upregulated PDX1 expression [74]. Although insulin content did not increase in culture, ICC transplanted under the kidney capsule were found to give rise to grafts that had significantly higher levels of insulin compared to adult islets and completely reversed diabetes in STZ-treated nude rats.
In another study, nicotinamide treatment induced maturation of human fetal pancreatic islet cells, but not adult β-cells, and resulted in otherwise unresponsive glucose-stimulated insulin release [75]. Although fetal β-cells failed to proceed through the differentiation process to achieve a terminal phenotype in culture, this finding suggests that they are able to go through the maturation process in vivo. Taken together, these tissue culture models have provided groundwork for further studies to dissect the molecular mechanisms of islet cell differentiation from developmentally earlier multipotent or pluripotent stem cells such as ES cells.
Embryonic stem cells and β-cells
Derivation of embryonic stem (ES) cells and induced pluripotent stem (iPS) cells
Apart from the derivation of β-cells from various pancreatic stem cells in adult tissues and pancreas, pluripotent ES cells are another source to search for renewable β-cells. Mouse and human ES cells are derived from the inner cell mass of pre-implantation blastocysts [76, 77], but they require different signaling pathways to maintain pluripotency. Recently, cells from epiblast of post-implantation mouse and rat embryos have also been shown to give rise to pluripotent stem cells. These epiblast stem cells share many features with human embryonic stem cells (HESC), and differ from mouse embryonic stem cells (MESC), suggesting that the differences between mouse and human ES cells are due to their relationship to different developmental stages, rather than distinct species differences in the control of pluripotency [78].
Assuming that techniques were available for the efficient production of β-cells from HESC, there are still other factors restricting its widespread use for application in therapy. Besides ethical concerns, because of their derivation from human embryos, difficulties remain due to tissue rejection following transplantation. Much effort has been spent in the development of alternative methods of generating patient-specific stem cells. One way to circumvent the problem is to reprogram adult cells back to pluripotent ES-like cells. Takahashi et al. (2006 and 2007) derived these so-called induced pluripotent stem cells (iPSC) by transducing first mouse, and then human adult dermal fibroblasts using retroviruses carrying four stem cell factors, namely OCT4, SOX2, KLF4 and C-MYC [79, 80]. They later improved the protocol by eliminating C-MYC due to its function as an oncogene [81]. This potentially allows the generation of iPSC from patients with genetic disorders, providing a platform in which human diseases including diabetes mellitus may be studied in vitro [82], apart from the prospect of patient-specific cells that would obviate immune rejection in transplantation regimens.
β-cells differentiated from MESC by cell-trapping
In the first successful attempt to induce pancreatic differentiation from MESC, Soria et al. (2001) employed a cell-trapping system, followed by cell-lineage selection and maturation protocols to obtain insulin-producing clones [83]. The cells were transfected with a plasmid containing a neomycin resistance gene under the control of the insulin promoter, and hygromycin selection cassette driven by a constitutively active phosphoglycerate kinase (PGK) promoter. The hygromycin-resistant clones were cultured in suspension to induce embryoid body (EB) differentiation. Neomycin antibiotic pressure was applied later to select for cells expressing factors that activate the insulin promoter. In one clone, the cells released insulin in response to glucose, although the insulin content was low. These insulin-positive cells normalized hyperglycemia when transplanted into the spleen of STZ-induced diabetic mice. In these mice, blood glucose levels remained normal and insulin-positive cells were present in the liver and in the spleen even after 42 weeks of transplantation [84].
Several genes, such as nkx6.1, bind to the insulin promoter and regulate insulin transcription in β-cells. It was reasoned that insulin-producing cells could be enriched if cells that activated its upstream promoters were isolated and expanded. Hence, the aforementioned approach was refined by replacing the insulin promoter with the nkx6.1 promoter to allow the selection of cells expressing pro-endocrine genes that have an activated nkx6.1 gene [85]. The hygromycin-resistant clones were first differentiated as EB in the presence of several exogenous agents. Treatment with nicotinamide, anti-Shh, and co-culture EB in the presence of pancreatic rudiments of E17.5 mice resulted in optimal nkx6.1 expression. An almost pure population of insulin-positive cells was produced, of which 20% were Pdx1 co-expressing cells from several clones. Subsequent transplantation of these cells into STZ-induced diabetic mice normalized their glycemia. The same group later demonstrated that substitution of insulin for pancreas-conditioned medium did not result in differentiated β-cells, suggesting that the differentiation effect was mostly due to soluble factors released by the pancreatic rudiment [86].
β-cells differentiated through neural progenitors from MESC
Pancreas organogenesis shares similarities with the development of the nervous system, despite pancreas and CNS originating from different germ layers [16, 87, 88]. Pancreatic endocrine cells also share several characteristics with neurons, for instance, islet cells are electrically excitable [38, 87]. Additionally, insulin-producing cells have also been observed in the invertebrate nervous system [89-91] and in primary cultures of mammalian fetal brain [92]. Hence, many protocols for deriving β-cells from MESC were designed first to produce or select for neural progenitors defined by nestin expression [93] and then to direct pancreatic islet differentiation in subsequent steps. As described in the previous section, nestin has been detected in a small population of pancreatic cells that have been proposed as possible islet precursors [59, 87].
Lumelsky et al. (2001) demonstrated that a small proportion of insulin-producing cells could be derived from MESC with five sequential in vitro differentiation steps, during which cultures were highly enriched in cells expressing nestin [94]. Nestin-positive cells derived from EB were expanded in B27/N2 neurobasal medium supplemented first with bFGF and subsequently cultured in nicotinamide. The outcome of their protocol was the formation of cell clusters connected by a web of neurons, with 10-30% insulin-expressing cells localized inside these clusters. Although the differentiated cells were immunoreactive to insulin, RT-PCR revealed that insulin2, glut2 and pdx1 mRNA levels were low and β-cell-specific insulin1 mRNA was never detected. These insulin-positive cells contained 50 times less insulin than normal islet cells. Not surprisingly, with such small amounts of insulin, the cells failed to correct hyperglycemia when grafted into STZ-treated diabetic mice. Nonetheless, the transplanted cells underwent rapid vascularization. The grafted mice were able to maintain their body weight and survived for longer periods of time than hyperglycemic sham-grafted controls.
With minor modifications in Lumelsky’s differentiation protocols, populations of nestin-expressing MESC derivatives differentiated into insulin-producing cells showing increased levels of β-cell transcripts and insulin production. Hori et al. (2003) substituted the phosphoinositide 3-kinase (PI3K) inhibitor, LY294002 for B27 at the later stage of differentiation [95]. LY294002 has been shown to increase total endocrine cell number and insulin content from the human fetal pancreas [96], as well as to prevent neurite outgrowth from neuroendocrine cells [97]. This modification yielded insulin-producing cell clusters (IPCC) with morphological similarities to pancreatic islets. Immunocytochemistry staining showed that Pdx1, GLUT2 and glucokinase were co-expressed by insulin-positive cells. However, the cells were also distinct from islets in numerous ways; while 95-97% of the cells were insulin-positive, and 2-3% were glucagon-positive, no staining for somatostatin and PP was detected. Nonetheless, these IPCC increased circulating insulin levels, reduced weight loss, improved glycemic control and completely rescued the survival of diabetic mice. Intriguingly, when LY294002 was not used, transplanted cells formed tumors that resembled teratomas, suggesting that undifferentiated cells were still present.
The discrepancy in insulin1 mRNA and insulin protein expression, and the lack of C-peptide release in these two studies leads us towards the hypothesis that cells differentiated with the nestin protocol take up exogenous insulin from the media. By adding fluorescein-labeled insulin in the media, cells were found to absorb these insulin molecules, hence were immunoreactive to anti-insulin antibody. An explanation for this active insulin uptake is the prevalence of apoptosis and necrosis due to suboptimal culture condition. Although these cells release insulin in response to glucose, C-peptide release was never detected, suggesting that de novo insulin synthesis was not a common outcome of cells differentiated from nestin-expressing neuroendocrine progenitors [98, 99].
Later, Blyszczuk et al. (2004) reported that MESC can be induced to differentiate into insulin-producing cells without employing the 'nestin-selection' step [100]. In this study, the use of ITSF (insulin, transferrin, selenium and fibronectin) and bFGF were avoided after replating EB on monolayer, so that selection or enrichment of specific cell types before induction of pancreatic differentiation would not occur. Functional β-like cells were generated following pancreatic differentiation by serum-free medium containing nicotinamide and laminin. However, ITSF were still added in the later stage of the differentiation protocol to compensate for serum-free culture conditions. Although it is not clear whether the addition of ITSF to the medium would select for nestin-positive cells, they concluded that β-cell precursors transiently expressed nestin during differentiation in vitro.
Exogenous expression of β-cell transcription factors
Constitutive expression of transcription factors with a role in pancreatic development such as pdx1 or pax4 has also been used to enhance differentiation of insulin-producing β-cells from MESC [101]. During in vitro differentiation, important changes in expression levels of pancreatic genes were detected in Pax4-positive cells, and to a lesser extent, in Pdx1-positive cells. IAPP, glut2 and insulin transcripts were upregulated in Pax4-positive cells at a later stage of differentiation, although ngn3 expression remained constant [101]. There was also a significant increase in the number of nestin-expressing cells in Pax4-positive cells, and this increase was corroborated by an increased level of intracellular insulin. As expected, no significant differences in glucagon-expressing cells were observed. Pax4-positive cells were then cultivated in Spinner rotation cultures to promote their histotypic maturation into spheroid islet-like clusters grown in suspension. Electron microscopy detected insulin-labeled secretory granules in Pax4-derived cells, and notably, these cells were able to normalize blood glucose levels in STZ-treated diabetic mice.
Inducible expression of Pdx1 in MESC also resulted in increased expression of pancreatic endocrine transcription factors during EB differentiation [102]. A MESC line, in which exogenous Pdx1 expression was regulated by a tet-off system integrated into the ROSA26 locus, was employed to examine the effect of Pdx1 expression during in vitro differentiation using a protocol similar to Lumelsky's protocol. For differentiation, cells were grown in the absence of doxycycline to induce the expression of Pdx1 and incubated with the addition of B27/N2, nicotinamide, KGF and EGF. The results showed that Pdx1 induction enhanced the expression of the insulin, somatostatin, Kir6.2, glucokinase, ngn3, p48 and pax6 genes. An overview of the methods used to generate β-cells from MESC is illustrated in Figure 1.
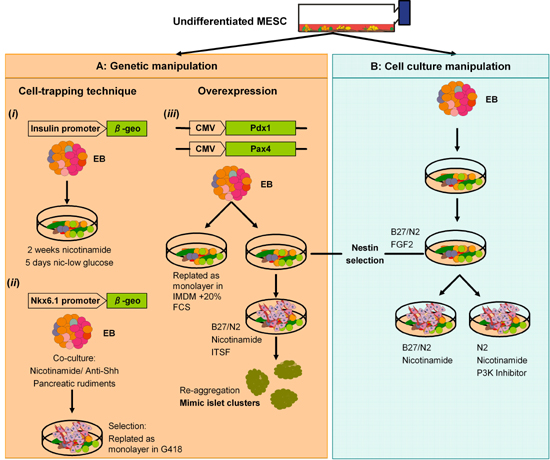 |
 |
Figure 1. In vitro differentiation of putative β-cells from mouse embryonic stem cells (MESC). A: Genetic manipulation. i: Soria et al. (2001) used a 'cell-trapping' protocol to select for insulin-producing cells expressing the β-geo gene under the control of the human Insulin gene promoter [84]. This strategy was later refined by placing the β-geo gene under the control of the promoter of Nkx6.1 (ii). iii: Enforced expression of Pax4 or Pdx1 increased the frequency with which insulin-producing cells were isolated from differentiating MESC. B: Cell culture manipulation of ES cells. Lumelsky et al. (2001) developed a five-step protocol based on methods known to promote the generation of neural cell types from MESC [94]. Nestin-expressing cells were cultured in B27/N2 neurobasal medium went on to form cell clusters. Although the nature of insulin-staining cells derived by this method remains controversial, other groups have successfully used variations on this procedure to generate similar cell types. EB: embryoid bodies. |
|
Early progress with human ES cells (HESC)
Expression of genes characteristically involved in the establishment of the mouse embryonic pancreas, including NGN3 and PDX1 transcription factors as well as INSULIN, GCK and GLUT2 were detected by RT-PCR analysis in HESC cultures during EB and monolayer culture differentiation [103]. Immunohistochemistry analysis revealed that 60-70% EB contained insulin-expressing cells, although only up to 2% of cells in an EB stained for insulin. Not surprisingly, with such low numbers of insulin-positive cells, further characterizations were impossible. Furthermore, glucose-responsiveness of these insulin-positive EB could not be determined.
Three years after the first report with MESC was published, it was shown that HESC could be induced to form insulin-producing islet-like clusters similar to immature pancreatic β-cells [104]. The differentiation strategy in this study was a modification of the 'nestin-selection' method. Similarly, HESC were differentiated in EB and replated on monolayer to allow the growth of nestin-positive neuroendocrine progenitors. Similar to MESC, differentiated cells expressed low insulin. However, by reducing the glucose concentration in later stages of differentiation and by reaggregating these clusters in suspension, insulin expression and secretion was increased. Insulin-producing clusters produced by this method resemble immature pancreatic embryonic cells. A substantial number of cluster cells were co-stained for insulin and glucagon or somatostatin. RT-PCR detects the expression of immature islet genes such as NGN3 but low expression of ISLET1, PDX1 and GCK. Although they showed a response to the different β-cell agonists and antagonists, their responsiveness to glucose was minimal. According to our experience, nestin is expressed by undifferentiated HESC and its expression remained constant during differentiation (Figure 2). Thus, the protocol to differentiate β-cells using nestin selection should be reevaluated considerably.
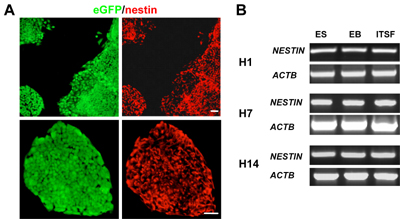 |
|
Figure 2. Nestin expression in human embryonic stem cells (HESC). A: Fluorescent microscopy showing that HESC colonies were immunoreactive to nestin. B: RT-PCR analysis of NESTIN expression in H1, H7 and H14 HESC lines during in vitro differentiation. NESTIN transcripts were detected in undifferentiated stem cells and embroid bodies (EB). There was no significant difference in the levels of NESTIN expression when the cells were grown in ITSF serum-free medium, indicating that ITSF medium did not increase the expression of NESTIN in HESC. Scale bars: 50µm. |
|
Co-culture of HESC with mouse embryonic pancreas
Given the inefficiency and controversies for deriving β-cells from nestin-expressing neuroendocrine progenitors, a more plausible approach is to direct ES cells through a process that mimics normal pancreas development. HESC grown to confluency gave rise to spontaneously differentiated cells that yielded FOXA2 and PDX1 co-expressing definitive endoderm (DE) cells at the periphery of the colonies. These early pancreatic progenitors were manually picked and co-transplanted with E11.5 mouse embryonic dorsal pancreas under the kidney capsule of SCID mouse. Eight weeks after transplantation, clusters consisted mainly of proinsulin-expressing β-like cells and a few glucagon and amylase-secreting cells were detected in the grafted section. Intriguingly, transplanting DE cells alone did not result in the appearance of β-like cells. This finding suggests that the stimulatory cues from co-grafted pancreas and an in vivo milieu were necessary to coax HESC to differentiate into β-cells.
β-cells from HESC-derived definitive endoderm cells
Studies with MESC revealed that by restricting serum in the presence of activin A in media induced the formation of Bracyhury-expressing mesendoderm cells in EB, from which endoderm cells may develop later [15]. D'Armour modified this protocol by inducing HESC to differentiate in monolayer in the presence of 100ng/ml activin A and low serum [105]. 80% of cells in activin A-treated cultures were immunoreactive to FOXA2 and SOX17, suggesting the enrichment of DE cells. High activin/nodal, but reduced insulin/insulin-like growth factor (IGF) signaling and suppression of PI3K-dependent pathway, are critical for the development of HESC to DE cells [106].
These DE cells can further differentiate into pancreatic endocrine cells in the presence of Hedgehog- and Notch-signaling inhibitors [107]. In brief, KAAD-cyclopamine and FGF10 were used to promote endoderm patterning and proliferation from DE cells. The outcome of this stage was the generation of PDX1-expressing gut-tube endodermal like cells. These cells were subsequently cultured in retinoic acid (RA), α-secretase inhibitor and exendin-4 to promote the transition towards hormone-expressing pancreatic endocrine cells. This protocol generated 7-12% of insulin-positive cells, and similar to fetal β-cells, they released C-peptide in response to depolarizing reagents such as potassium chloride (KCl) and tolbutamide. Nevertheless, there was only minimal glucose-induced C-peptide release. With a slight modification to this protocol, Jiang et al. (2007) generated islet-like clusters from feeder free HESC-derived DE cells in low adherent cultures to form aggregates in the presence of noggins and nicotinamide [107]. Similarly, these aggregates produced cells secreting various pancreatic endocrine hormones, and notably, released C-peptide in response to glucose. The use of xeno-free HESC will be prerequisite for utilizing these cells in stem cell therapies [108, 109].
It is reasoned that in vivo maturation might be necessary for the functional maturation of HESC-derived pancreatic endocrine or β-cells. Pancreatic endocrine cells, owing to their precursor status, may be more potent than β-cells in undergoing maturation should they respond to in vivo milieu. Harvested pancreatic endocrine cells went on to form aggregates on gelatin foam sponges and matrigel overlays. These cells were then harvested and grafted into STZ-treated immuno-compromised mice. Although no significant level of human C-peptide was detected within the first month, long term engraftment data revealed that glucose-stimulated serum levels of human C-peptide increased rapidly during the next sixty days [110]. Nonetheless, because purification steps were not used, teratomas containing various other tissues of ectodermal and mesodermal were formed in most of the animals. Thus, future improvement will require purification of pancreatic cell types and depletion of inappropriate cell types prior to transplantation.
β-cells isolated from Pax4-expressing HESC
Our own laboratory has described an efficient method to enhance β-cell differentiation from HESC [111]. We first overexpressed Pax4 in HESC from a constitutively active CAG (human cytomegalovirus major immediate early enhancer- chicken β-actin- rabbit β-globin) hybrid promoter and induced differentiation in EB model. We reasoned that a proportion of DE cells spontaneously differentiated from EB, substantiated by the detection of FOXA2 and NEUROD1 transcripts. Various key genes expressed in mature β-cells such as prohormone convertase 1/3 (PC1/3), GCK and GLUT2 were upregulated in Pax4-expressing EB. These cells also expressed markers of voltage-gated Ca2+-channels (VGCC) at comparable levels to human fetal pancreas. In addition, the increase in VGCC genes correlated with an enhanced responsiveness of EB to muscarinic receptor agonists such as KCl. When replated on monolayer, EB went on to form putative β-cells. These cells could be isolated by fluorescence activated cell sorting (FACS) using Newport Green dye based on the high zinc content in insulin-containing cells. Purified β-like cells were enriched in PDX1 and INSULIN transcripts but depleted in OCT4 expression, suggesting the absence of non-pancreatic cell types and residual undifferentiated cells. Isolated cells also synthesized proinsulin, expressed C-peptide and responded to tolbutamide. Whereas this study shows the possibility to increase the differentiation of β-cells from HESC, further work is required to characterize in vivo maturation and function of these cells. A summary of the methods used to generate β-cells from HESC is illustrated in Figure 3.
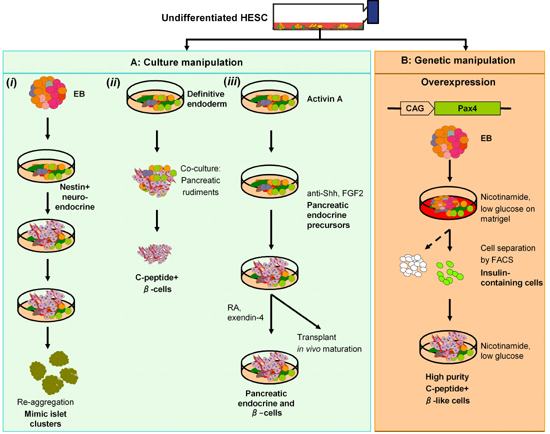 |
|
Figure 3. Overview of studies on generation of putative β-cells from HESC. A: Cell culture manipulation. i: Nestin-expressing cells from EB, similar to those differentiated from MESCs gave rise to insulin-immunoreactive cells. Cells were re-aggregated in suspension cultures to yield islet-like clusters consisted of cells expressing pancreatic endocrine hormones. ii: Definitive endoderm (DE) cells differentiated at the edge of HESC colonies were manually picked, co-transplanted with mouse embryonic pancreas further differentiated to β-cells in vivo. iii: A 2D culture generated DE cells by activin A treatment. DE cells responded to hedgehog and Notch inhibitors, giving rise to cells that express pancreas endocrine hormones. Cells transplanted in STZ-induced mice ameliorated hyperglycemia. B: β-like cells were enriched from Pax4-expressing EB and replated on monolayer with nicotinamide and low glucose. A zinc chelating dye was used to isolate insulin-containing cells. These cells went on to form C-peptide-positive cells that responded to β-cell secretagogues. It remains to be determined whether these isolated β-cells rescue hyperglycemia in a mouse model. |
|
Generally, the formation of a β-cell from a pluripotent stem cell represents a part of the complex choreography of embryonic development. If the formation of a β-cell in vivo is to be strictly followed, expression of key regulatory genes for this sequential commitment should be regulated in a time-dependent manner. A major challenge is that ES cells support the early stages of endoderm and pancreatic endocrine cell formation in vitro but do not robustly generate glucose-responsive β-cells, making diabetes treatment using stem cell therapy untenable. Inefficiencies in deriving a scalable source of β-cells from HESC in vitro likely reflect the inability of undifferentiated stem cells to mimic these normally intricate in vivo gene expression patterns. Furthermore, published studies so far revealed that HESC-derived β-cells were not glucose-responsive, but acquired further maturation once transplanted into mice. These findings add support to the contention that these cells mimic β-cells in the human fetal pancreas. In order to recapitulate pancreas organogenesis in vitro, an improved three-dimensional EB differentiation model and manipulation of extrinsic signals may be employed to support the formation of pancreatic mesenchyme, acini, islets and β-cells in a spatially and temporally controlled pattern.
Future prospects of stem cell therapy using HESC- or iPSC-derived β-cells
Several findings have underscored the necessity for deriving more HESC for research purposes. As HESC lines are derived from a genetically heterogenous population, their biological variations, heterogeneity, genetic and epigenetic differences contribute to the variations in developmental potential. Various studies have reported that HESC lines exhibited marked differences in differentiation propensity into specific lineages [112, 113]. Notably, genes that play the key role in maintaining pluripotency, such as OCT4, NANOG and CRIPTO showed little variation; however, marker expression of multiple tissue-specific lineages became variable after the cells began to differentiate [114]. Some lines exhibited a marked potential to differentiate into one lineage, often with more than 100-fold differences in lineage-specific gene expression. When gene expression levels of the same HESC lines with different passage numbers were compared, consistent patterns within the same line but significant variations between lines remained. This observation argues against the possibility that variability of culture technique or senescence of HESC contributed to marked differences in differentiation propensity in different lines. We have also observed variations of pancreatic lineage differentiation potential from different HESC lines (Figure 4). Thus, it would be advantageous to work with HESC that are inclined toward pancreatic differentiation.
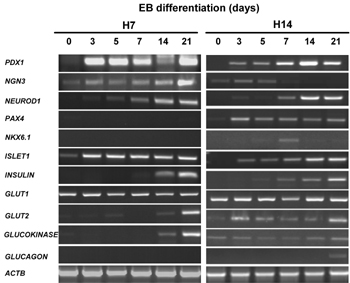 |
|
Figure 4. RT-PCR analysis of β-cell gene expression in two different HESC lines (H7 and H14) during in vitro differentiation. 0-day cells are undifferentiated stem cells. Embryoid bodies (EB) of different stages were collected for RNA extraction. There were transient expressions of NGN3, PAX4 and NKX6.1 transcription factors at early stages of H14 EB differentiation. However, the expressions of PAX4 and NKX6.1 in H7 EB were minimal. Up-regulation of NEUROD1 and ISLET1 transcripts was detected in the course of EB differentiation. Differentiated EB expressed INSULIN, GLUT2 and GLUCOKINASE β-cell markers at a later stage of EB differentiation, suggesting that H7 and H14 cells were able to produce β-cells in the course of spontaneous differentiation. There was no change in GLUT1 transcript levels during EB differentiation. GLUCAGON transcripts were only detected in H14 EB. |
|
The derivation of iPSC opens new possibilities to reprogram pluripotent stem cell lines from individual patients to avoid immunological rejections in cell therapy. However, time and costs necessary for derivation, differentiation and safety testing of good manufacturing practice (GMP)- or clinical-grade iPSC from each individual may exceed practical and economical limits. Another concern is the age of the patient; iPSC obtained from young adults are healthier than those derived from mature adults.
An attractive alternative to eliminate the age concern and to reduce the likelihood of graft rejection in stem cell therapy is through the use of human leukocyte antigen (HLA)-haplotype cell banking, from which a best match could be selected. The results of pancreas transplantation will also be improved by minimizing HLA mismatches [115]. Recently, Taylor et al. (2005) estimated the required number of HESC lines for beneficial HLA-matching in the UK. They proposed that about 150 HESC lines would be needed for most of the UK population, and that as few as ten might be sufficient if one were to prospectively identify cell lines that could serve a larger number of patients, such as lines homozygous for common HLA types [116, 117]. Based on estimates using Japanese population, Nakatsuji et al. (2006) also estimated that with a bank of HESC lines from 170 randomly selected donated embryos, 80% of the patients could be expected to find at least one HESC line with only a single mismatch at one HLA locus or even a better match [118]. Thus, the production of HLA-mapped clinically-safe iPSC would greatly minimize the ethical concern and enable the creation of more efficient HLA-haplotype banks. Therefore, in cases when stem cells cannot be obtained from the patient themselves (e.g., in an aged patient), iPSC lines derived from an unrelated individual may eventually become alternative sources of various cell types for transplantation therapy.
However, whether cell transplantation will ultimately be the approach of choice for regenerative medicine is a moot point. Many technical challenges remain to be overcome, and it is possible that a better understanding of the basic biology of the pancreas, from the point of view of both its development and subsequent homeostasis, may offer insight that will provide alternative approaches to curing diabetes and related diseases. Studies of ES cells and other stem cells may well play a substantial role in acquiring the necessary knowledge, and the development of patient-specific iPSC certainly offers a new approach to advancing disease models as well as tools for screening new drugs that could play a role in novel treatments in the future.
Acknowledgments:
The authors are grateful to Juvenile Diabetes Research Foundation (JDRF) for funding some of the work presented in this article. Chee Liew is a recipient of California Institute of Regenerative Medicine (CIRM) Postdoctoral Fellowship.
References
- Yki-Jarvinen H, Koivisto V. Natural course of insulin resistance in type I diabetes. N Engl J Med 1986. 315:224-230. [DOD]
- Yki-Jarvinen HD, Mott AA, Young KS, Bogardus C. Regulation of glycogen synthase and phosphorylase activities by glucose, insulin and basal enzyme activity in human skeletal muscle. J Clin Invest 1987. 80:95-100. [DOD] [CrossRef]
- Peltoniemi P, Yki-Jarvinen H, Oikonen V, Oksanen A, Takala TO, Ronnemaa T, Erkinjuntti M, Knuuti MJ, Nuutila P. Resistance to exercise-induced increase in glucose uptake during hyperinsulinemia in insulin-resistant skeletal muscle of patients with type 1 diabetes. Diabetes 2001. 50:1371-1377. [DOD] [CrossRef]
- Docherty K. Growth and development of the islets of Langerhans: implications for the treatment of diabetes mellitus. Curr Opin Pharmacol 2001. 1:641-649. [DOD] [CrossRef]
- Kaaja R, Rönnemaa T. Gestational diabetes: pathogenesis and consequences to mother and offspring. Rev Diabet Stud 2008. 5(4):194-202. [DOD] [CrossRef]
- Shapiro AM, Lakey J, Ryan E, Korbutt G, Toth E, Warnock G, Kneteman N, Rajotte R. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N Engl J Med 2000. 343:230-238. [DOD] [CrossRef]
- Shapiro AM, Ricordi C, Hering BJ, Auchincloss H, Lindblad R, Robertson RP, Secchi A, Brendel MD, Berney T, Brennan DC, et al. International Trial of the Edmonton Protocol for Islet Transplantation. N Engl J Med 2006. 355:1318-1330. [DOD] [CrossRef]
- Berney T, Toso C. Monitoring of the islet graft. Diabetes Metab 2006. 32:503-512. [DOD]
- Xu G, Stoffers DA, Habener JF, Bonner-Weir S. Exendin-4 stimulates both beta-cell replication and neogenesis, resulting in increased beta-cell mass and improved glucose tolerance in diabetic rats. Diabetes 1999. 48:2270-2276. [DOD] [CrossRef]
- Street CN, Lakey JR, Rajotte RV, Shapiro AM, Kieffer TJ, Lyon JG, Kin T, Korbutt GS. Enriched human pancreatic ductal cultures obtained from selective death of acinar cells express pancreatic and duodenal homeobox gene-1 age-dependently. Rev Diabet Stud 2004. 1(2):66-79. [DOD] [CrossRef]
- Bonner-Weir S, Baxter L, Schuppin G, Smith F. A second pathway for regeneration of adult exocrine and endocrine pancreas. A possible recapitulation of embryonic development. Diabetes 1993. 42:1715-1720. [DOD] [CrossRef]
- Bouwens L, Lu WG, De Krijger R. Proliferation and differentiation in the human fetal endocrine pancreas. Diabetologia 1997. 40(4):398-404. [DOD] [CrossRef]
- Tyrberg B, Ustinov J, Otonkoski T, Andersson A. Stimulated endocrine cell proliferation and differentiation in transplanted human pancreatic islets: effects of the ob gene and compensatory growth of the implantation organ. Diabetes 2001. 50:301-307. [DOD] [CrossRef]
- Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 2004. 429:41-46. [DOD] [CrossRef]
- Kubo A, Shinozaki K, Shannon JM, Kouskoff V, Kennedy M, Woo S, Fehling HJ, Keller G. Development of definitive endoderm from embryonic stem cells in culture. Development 2004. 131:1651-1662. [DOD] [CrossRef]
- Kim SK, Hebrok M. Intercellular signals regulating pancreas development and function. Genes Dev 2001. 15(2):111-127. [DOD] [CrossRef]
- Richardson M, Hanken J, Gooneratne M, Pieau C, Raynaud A, Selwood L, Wright G. There is no highly conserved embryonic stage in the vertebrates: implications for current theories of evolution and development. Anat Embryol 1997. 196(2):91-106. [DOD] [CrossRef]
- Polak M, Bouchareb-Banaei L, Scharfmann R, Czernichow P. Early pattern of differentiation in the human pancreas. Diabetes 2000. 49:225-232. [DOD] [CrossRef]
- Stefan Y, Grasso S, Perrelet A, Orci L. A quantitative immunofluorescent study of the endocrine cell populations in the developing human pancreas. Diabetes 1983. 32:293-301. [DOD] [CrossRef]
- Clark A, Grant AM. Quantitative morphology of endocrine cells in human fetal pancreas. Diabetologia 1983. 25:31-35. [DOD]
- Piper K, Ball SG, Keeling JW, Mansoor S, Wilson DI, Hanley NA. Novel SOX9 expression during human pancreas development correlates to abnormalities in Campomelic dysplasia. Mech Dev 2002. 116(1-2):223-226. [DOD] [CrossRef]
- Piper K, Brickwood S, Turnpenny LW, Cameron IT, Ball SG, Wilson DI, Hanley NA. Beta cell differentiation during early human pancreas development. J Endocrinol 2004. 181:11-23. [DOD] [CrossRef]
- Beattie G, Butler C, Hayek A. Morphology and function of cultured human fetal pancreatic cells transplanted into athymic mice: a longitudinal study. Cell Transplant 1994. 3:421-425. [DOD]
- Seymour PA, Freude KK, Tran MN, Mayes EE, Jensen J, Kist R, Scherer G, Sander M. SOX9 is required for maintenance of the pancreatic progenitor cell pool. Proc Natl Acad Sci USA 2007. 104(6):1865-1870. [DOD] [CrossRef]
- Gradwohl G, Dierich A, LeMeur M, Guillemot F. neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci USA 2000. 97(4):1607-1611. [DOD] [CrossRef]
- Naya F, Huang H, Qiu Y, Mutoh H, DeMayo F, Leiter A, Tsai M. Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/neuroD-deficient mice. Genes Dev 1997. 11:2323-2334. [DOD] [CrossRef]
- Malecki MT, Jhala US, Antonellis A, Fields L, Doria A, Orban T, Saad M, Warram JH, Montminy M. Mutations in NEUROD1 are associated with the development of type 2 diabetes mellitus. Nat Genet 1999. 23(3):323-328. [DOD] [CrossRef]
- Mansouri A, St-Onge L, Gruss P. Role of genes in endoderm-derived organs. Trends Endocrinol Metab 1999. 10(4):164-167. [DOD] [CrossRef]
- Dohrmann C, Gruss P, Lemaire L. Pax genes and the differentiation of hormone-producing endocrine cells in the pancreas. Mech Dev 2000. 92(1):47-54. [DOD] [CrossRef]
- Hill M, Asa S, Drucker D. Essential role for Pax6 in control of enteroendocrine proglucagon gene transcription. Mol Endocrinol 1999. 13(9):1474-1486. [DOD] [CrossRef]
- St-Onge L, Sosa-Pineda S, Chowdhury K, Mansouri A, Gruss P. Pax6 is required for differentiation of glucagon-producing alpha-cells in mouse pancreas. Nature 1997. 387:406-409. [DOD] [CrossRef]
- Smith SB, Ee HC, Conners JR, German MS. Paired-homeodomain transcription factor PAX4 acts as a transcriptional repressor in early pancreatic development. Mol Cell Biol 1999. 19(12):8272-8280. [DOD]
- Sosa-Pineda B, Chowdhury K, Torres M, Oliver G, Gruss P. The Pax4 gene is essential for differentiation of insulin-producing â cells in the mammalian pancreas. Nature 1997. 386,:399-402. [DOD] [CrossRef]
- Matsuoka TA, Artner I, Henderson E, Means A, Sander M, Stein R. The MafA transcription factor appears to be responsible for tissue-specific expression of insulin. Proc Natl Acad Sci USA 2004. 101(9):2930-2933. [DOD] [CrossRef]
- Jensen J, Serup P, Karlsen C, Nielsen T, Madsen O. mRNA profiling of rat islet tumors reveals nkx 6.1 as a beta-cell-specific homeodomain transcription factor. J Biol Chem 1996. 271:18749-18758. [DOD] [CrossRef]
- Rudnick A, Ling TY, Odagiri H, Rutter WJ, German MS. Pancreatic beta cells express a diverse set of homeobox genes. Proc Natl Acad Sci USA 1994. 91(25):12203-12207. [DOD]
- Olbrot M, Rud J, Moss LG, Sharma A. Identification of beta-cell-specific insulin gene transcription factor RIPE3b1 as mammalian MafA. Proc Natl Acad Sci USA 2002. 99:6737-6742. [DOD] [CrossRef]
- Edlund H. Developmental biology of the pancreas. Diabetes 2001. 50:S5-S9. [DOD] [CrossRef]
- Hui H, Perfetti R. Pancreas duodenum homeobox-1 regulates pancreas development during embryogenesis and islet cell function in adulthood. Eur J Endocrinol 2002. 146:129-141. [DOD] [CrossRef]
- Stoffers DA, Zinkin NT, Stanojevic V, Clarke WL, Habener JF. Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat Genet 1997. 15:106-110. [DOD] [CrossRef]
- Stoffers D, Ferrer J, Clarke W, Habener J. Early-onset type-II diabetes mellitus (MODY4) linked to IPF1. Nat Genet 1997. 17:138-139. [DOD] [CrossRef]
- Hattersley AT. Maturity-onset diabetes of the young: clinical heterogeneity explained by genetic heterogeneity. Diabet Med 1998. 15(1):15-24. [DOD] [CrossRef]
- Shiroi A, Yoshikawa M, Yokota H, Fukui H, Ishizaka S, Tatsumi K, Takahashi Y. Identification of insulin-producing cells derived from embryonic stem cells by zinc-chelating dithizone. Stem Cells 2002. 20:284-292. [DOD] [CrossRef]
- Lukowiak B, Vandewalle B, Riachy R, Kerr-Conte J, Gmyr V, Belaich S, Lefebvre J, Pattou F. Identification and purification of functional human beta-cells by a new specific zinc-fluorescent probe. J Histochem Cytochem 2001. 49(4):519-528. [DOD]
- Chausmer AB. Zinc, insulin and diabetes. J Am Coll Nutr 1998. 17:109-115. [DOD]
- Dodson G, Steiner D. The role of assembly in insulin’s biosynthesis. Curr Opin Struct Biol 1998. 8(2):189-194. [DOD] [CrossRef]
- Guillam M, Hummler E, Schaerer E, Wu J, Birnbaum M, Beermann F, Schmidt A, Deriaz N, Thorens B. Early diabetes and abnormal postnatal pancreatic islet development in mice lacking Glut-2. Nat Genet 1997. 17:327-330. [DOD] [CrossRef]
- Thorens B, Guillam MT, Beermann F, Burcelin R, Jaquet M. Transgenic reexpression of GLUT1 or GLUT2 in pancreatic beta cells rescues GLUT2-null mice from early death and restores normal glucose-stimulated insulin secretion. J Biol Chem 2000. 275:23751-23758. [DOD] [CrossRef]
- Guillam MT, Dupraz P, Thorens B. Glucose uptake, utilization, and signaling in GLUT2-null islets. Diabetes 2000. 49(9):1485-1491. [DOD] [CrossRef]
- Hui H, Wright C, Perfetti R. Glucagon-like peptide 1 induces differentiation of islet duodenal homeobox-1-positive pancreatic ductal cells into insulin-secreting cells. Diabetes 2001. 50:785-796. [DOD] [CrossRef]
- Molinete M, Irminger JC, Tooze SA, Halban PA. Trafficking/sorting and granule biogenesis in the beta-cell. Semin Cell Dev Biol 2000. 11(4):243-251. [DOD] [CrossRef]
- Rosenberg L. In vivo cell transformation: neogenesis of beta cells from pancreatic ductal cells. Cell Transplant 1995. 4:371-383. [DOD] [CrossRef]
- Goudswaard WB, Houthoff HJ, Koudstaal J, Zwierstra RP. Nesidioblastosis and endocrine hyperplasia of the pancreas: a secondary phenomenon. Hum Pathol 1986. 17(1):46-54. [DOD] [CrossRef]
- Efrat S. Ex-vivo expansion of adult human pancreatic beta-cells. Rev Diabet Stud 2008. 5(2):116-122. [DOD] [CrossRef]
- Ramiya VK, Maraist M, Arfors KE, Schatz DA, Peck AB. Reversal of insulin-dependent diabetes using islets generated in vitro from pancreatic stem cells. Nat Med 2000. 6(3):278-282. [DOD] [CrossRef]
- Teitelman G, Alpert S, Polak JM, Martinez A, Hanahan D. Precursor cells of mouse endocrine pancreas coexpress insulin, glucagon and the neuronal proteins tyrosine hydroxylase and neuropeptide Y, but not pancreatic polypeptide. Development 1993. 118(4):1031-1039. [DOD]
- Bonner-Weir S, Taneja M, Weir GC, Tatarkiewicz K, Song KH, Sharma A, O'Neil JJ. In vitro cultivation of human islets from expanded ductal tissue. Proc Natl Acad Sci U S A 2000. 97(14):7999-8004. [DOD] [CrossRef]
- Xu X, D'Hoker J, Stange G, Bonne S, De Leu N, Xiao X, Van De Casteele M, Mellitzer G, Ling Z, Pipeleers D, Bouwens L, Scharfmann R, Gradwohl G, Heimberg H. Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell 2008. 132(2):197-207. [DOD] [CrossRef]
- Zulewski H, Abraham EJ, Gerlach MJ, Daniel PB, Moritz W, Muller B, Vallejo M, Thomas MK, Habener JF. Multipotential nestin-positive stem cells isolated from adult pancreatic islets differentiate ex vivo into pancreatic endocrine, exocrine, and hepatic phenotypes. Diabetes 2001. 50:521-533. [DOD] [CrossRef]
- Selander L, Edlund H. Nestin is expressed in mesenchymal and not epithelial cells of the developing mouse pancreas. Mech Dev 2002. 113(2):189-192. [DOD] [CrossRef]
- Klein T, Ling Z, Heimberg H, Madsen OD, Heller RS, Serup P. Nestin is expressed in vascular endothelial cells in the adult human pancreas. J Histochem Cytochem 2003. 51:697-706. [DOD]
- Lardon J, Rooman I, Bouwens L. Nestin expression in pancreatic stellate cells and angiogenic endothelial cells. Histochem Cell Biol 2002. 117(6):535-540. [DOD] [CrossRef]
- Gmyr V, Kerr-Conte J, Belaich S, Vandewalle B, Leteurtre E, Vantyghem M, Lecomte-Houcke M, Proye C, Lefebvre J, Pattou F. Adult human cytokeratin 19-positive cells reexpress insulin promoter factor 1 in vitro: further evidence for pluripotent pancreatic stem cells in humans. Diabetes 2000. 49:1671-1680. [DOD] [CrossRef]
- Strobel O, Dor Y, Stirman A, Trainor A, Fernandez-del Castillo C, Warshaw AL, Thayer SP. Beta-cell transdifferentiation does not contribute to preneoplastic/metaplastic ductal lesions of the pancreas by genetic lineage tracing in vivo. Proc Natl Acad Sci USA 2007. 104(11):4419-4424. [DOD] [CrossRef]
- Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature 2008. 455:627-632. [DOD] [CrossRef]
- Tosh D, Slack J. How cells change their phenotype. Nat Rev Mol Cell Biol 2002. 3(3):187-194. [DOD] [CrossRef]
- Kocher AA, Schuster MD, Szabolcs MJ, Takuma S, Burkhoff D, Wang J, Homma S, Edwards NM, Itescu S. Neovascularization of ischemic myocardium by human bone-marrow-derived antioblasts prevents cardiomyocyte apoptosis, reduces remodeling and improves cardiac function. Nat Med 2001. 7(4):430-436. [DOD] [CrossRef]
- Colter DC, Sekiya I, Prockop DJ. Identification of a subpopulation of rapidly self-renewing and multipotential adult stem cells in colonies of human marrow stromal cells. Proc Natl Acad Sci USA 2001. 98:7841-7845. [DOD] [CrossRef]
- Woodbury D, Schwarz EJ, Prockop DJ, Black IB. Adult rat and human bone marrow stromal cells differentiate into neurons. J Neurosci Res 2000. 61(4):364-370. [DOD] [CrossRef]
- Lechner A, Yang YG, Blacken RA, Wang L, Nolan AL, Habener JF. No evidence for significant transdifferentiation of bone marrow into pancreatic beta-cells in vivo. Diabetes 2004. 53(3):616-623. [DOD] [CrossRef]
- Ferber S, Halkin A, Cohen H, Ber I, Einav Y, Goldberg I, Barshack I, Seijffers R, Kopolovic J, Kaiser N, Karasik A. Pancreatice and duodenal homeobox gene 1 induces expression of insulin genes in liver and ameliorates streptozotocin-induced hyperglycemia. Nat Med 2000. 6(5):568-572. [DOD] [CrossRef]
- Ber I, Shternhall K, Perl S, Ohanuna Z, Goldberg I, Barshack I, Benvenisti-Zarum L, Meivar-Levy I, Ferber S. Functional, persistent and extended liver to pancreas transdifferentiation. J Biol Chem 2003. 278:31950-31957. [DOD] [CrossRef]
- Beattie GM, Otonkoski T, Lopez AD, Hayek A. Functional beta-cell mass after transplantation of human foetal pancreatic cells: differentiation or proliferation? Diabetes 1997. 46:244-248. [DOD]
- Movassat J, Beattie GM, Lopez AD, Hayek A. Exendin 4 up-regulates expression of PDX 1 and hastens differentiation and maturation of human fetal pancreatic cells. J Clin Endocrinol Metab 2002. 87:4775-4781. [DOD] [CrossRef]
- Otonkoski T, Beattie GM, Mally MI, Ricordi C, Hayek A. Nicotinamide is a potent inducer of endocrine differentiation in cultured human fetal pancreatic cells. J Clin Invest 1993. 92(3):1459-1466. [DOD] [CrossRef]
- Evans M, Kaufman J. Establishment in culture of pluripotential cells from mouse embryos. Nature 1981. 292:154-156. [DOD] [CrossRef]
- Thomson J, Itskovitz-Eldor J, Shapiro S, Waknitz M, Swiergiel J, Marshall V, Jones J. Embryonic stem cell lines derived from human blastocysts. Science 1998. 282:1145-1147. [DOD] [CrossRef]
- Brons IG, Smithers LE, Trotter MW, Rugg-Gunn P, Sun B, Chuva de Sousa Lopes SM, Howlett SK, Clarkson A, Ahrlund-Richter L, Pedersen RA, Vallier L. Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature 2007. 448(7150):191-195. [DOD] [CrossRef]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006. 126:663-676. [DOD] [CrossRef]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007. 131:861-872. [DOD] [CrossRef]
- Nakagawa M, Koyanagi M, Tanabe K, Takahashi K, Ichisaka T, Aoi T, Okita K, Mochiduki Y, Takizawa N, Yamanaka S. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol 2008. 26(1):101-106. [DOD] [CrossRef]
- Park IH, Arora N, Huo H, Maherali N, Ahfeldt T, Shimamura A, Lensch MW, Cowan C, Hochedlinger K, Daley GQ. Disease-specific induced pluripotent stem cells. Cell 2008. 134(5):877-886. [DOD] [CrossRef]
- Soria B, Roche E, Berna G, Leon-Quinto T, Reig JA, Martin F. Insulin-secreting cells derived from embryonic stem cells normalize glycemia in streptozotocin-induced diabetic mice. Diabetes 2000. 49:157-162. [DOD] [CrossRef]
- Soria B. In-vitro differentiation of pancreatic beta-cells. Differentiation 2001. 68(4-5):205-219. [DOD] [CrossRef]
- Leon-Quinto T, Jones J, Skoudy A, Burcin M, Soria B. In vitro directed differentiation of mouse embryonic stem cells into insulin-producing cells. Diabetologia 2004. 47(8):1442-1451. [DOD] [CrossRef]
- Vaca P, Martin F, Vegara-Meseguer JM, Rovira JM, Berna G, Soria B. Induction of differentiation of embryonic stem cells into insulin-secreting cells by fetal soluble factors. Stem Cells 2006. 24:258-265. [DOD] [CrossRef]
- Hunziker E, Stein M. Nestin-expressing cells in the pancreatic islets of langerhans. Biochem Biophys Res Commun 2000. 271(1):116-119. [DOD] [CrossRef]
- Schwitzgebel VM, Scheel DW, Conners JR, Kalamaras J, Lee JE, Anderson DJ, Sussel L, Johnson JD, German MS. Expression of neurogenin3 reveals an islet cell precursor population in the pancreas. Development 2000. 127(16):3533-3542. [DOD]
- Brogiolo W, Stocker H, Ikeya T, Rintelen F, Fernandez R, Hafen E. An evolutionarily conserved function of the Drosophila insulin receptor and insulin-like peptides in growth control. Curr Biol 2001. 11(4):213-221. [DOD] [CrossRef]
- Rulifson EJ, Kim SK, Nusse R. Ablation of insulin-producing neurons in flies: Growth and diabetic phenotypes. Science 2002. 296:1118-1120. [DOD] [CrossRef]
- Smit A, Vreugdenhil E, Ebberink R, Geraerts W, Klootwijk J, Joosse J. Growth-controlling molluscan neurons produce the precursor of an insulin-related peptide. Nature 1998. 331:535-538. [DOD] [CrossRef]
- Clarke DW, Mudd L, Boyd FT Jr, Fields M, Raizada MK. Insulin is released from rat brain neuronal cells in culture. J Neurochem 1986. 47(3):831-836. [DOD]
- Okabe S, Forsberg-Nilsson K, Spiro AC, Segal M, McKay RD. Development of neuronal precursor cells and functional postmitotic neurons from embryonic stem cells in vitro. Mech Dev 1996. 59(1):89-102. [DOD] [CrossRef]
- Lumelsky N, Blondel O, Laeng P, Velasco I, Ravin R, McKay R. Differentiation of embryonic stem cells to insulin-secreting structures similar to pancreatic islets. Science 2001. 292:1389-1394. [DOD] [CrossRef]
- Hori Y, Rulifson IC, Tsai BC, Heit JJ, Cahoy JD, Kim SK. Growth inhibitors promote differentiation of insulin-producing tissue from embryonic stem cells. Proc Natl Acad Sci USA 2002. 99(25):16105-16110. [DOD] [CrossRef]
- Ptasznik A, Beattie GM, Mally MI, Cirulli V, Lopez A, Hayek A. Phosphatidylinositol 3-kinase is a negative regulator of cellular differentiation. J Cell Biol 1997. 137:1127-1136. [DOD] [CrossRef]
- Kimura K, Hattori S, Kabuyama Y, Shizawa Y, Takayanagi J, Nakamura S, Toki S, Matsuda Y, Onodera K, Fukui Y. Neurite outgrowth of PC12 cells is suppressed by wortmannin, an specific inhibitor of phosphatidylinositol 3-kinase. J Biol Chem 1994. 269:18961-18967. [DOD]
- Rajagopal J, Anderson WJ, Kume S, Martinez OI, Melton DA. Insulin staining of ES cell progeny from insulin uptake. Science 2003. 299:363. [DOD]
- Hansson M, Tonning A, Frandsen U, Petri A, Rajagopal J, Englund MC, Heller RS, Hakansson J, Fleckner J, Skold HN, Melton D, Semb H, Serup P. Artifactual insulin release from differentiated embryonic stem cells. Diabetes 2004. 53:2603-2609. [DOD] [CrossRef]
- Blyszczuk P, Asbrand C, Rozzo A, Kania G, St-Onge L, Rupnik M, Wobus AM. Embryonic stem cells differentiate into insulin-producing cells without selection of nestin-expressing cells. Int J Dev Biol 2004. 48(10):1095-1104. [DOD] [CrossRef]
- Blyszczuk P, Czyz J, Kania G, Wagner M, Roll U, St-Onge L, Wobus AM. Expression of Pax4 in embryonic stem cells promotes differentiation of nestin-positive progenitor and insulin-producing cells. Proc Natl Acad Sci USA 2003. 100:998-1003. [DOD] [CrossRef]
- Miyazaki S, Yamato E, Miyazaki J. Regulated expression of pdx-1 promotes in vitro differentiation of insulin-producing cells from embryonic stem cells. Diabetes 2004. 53(4):1030-1037. [DOD] [CrossRef]
- Assady S, Maor G, Amit M, Itskovitz-Eldor J, Skorecki KL, Tzukerman M. Insulin production by human embryonic stem cells. Diabetes 2001. 50:1691-1697. [DOD] [CrossRef]
- Segev H, Fishman B, Ziskind A, Shulman M, Itskovitz-Eldor J. Differentiation of human embryonic stem cells into insulin-producing clusters. Stem Cells 2004. 22:265-274. [DOD] [CrossRef]
- D'Amour KA, Agulnick AD, Eliazer S, Kelly OG, Kroon E, Baetge EE. Efficient differentiation of human embryonic stem cells to definitive endoderm. Nat Biotechnol 2005. 23:1534-1541. [DOD] [CrossRef]
- McLean AB, D'Amour KA, Jones KL, Krishnamoorthy M, Kulik MJ, Reynolds DM, Sheppard AM, Liu H, Xu Y, Baetge EE, Dalton S. Activin A efficiently specifies definitive endoderm from human embryonic stem cells only when phosphatidylinositol 3-kinase signaling is suppressed. Stem Cells 2007. 25(1):29-38. [DOD] [CrossRef]
- Jiang J, Au M, Lu K, Eshpeter A, Korbutt G, Fisk G, Majumdar AS. Generation of insulin-producing islet-like clusters from human embryonic stem cells. Stem Cells 2007. 25:1940-1953. [DOD] [CrossRef]
- Xu C, Inokuma MS, Denham J, Golds K, Kundu P, Gold JD, Carpenter MK. Feeder-free growth of undifferentiated human embryonic stem cells. Nat Biotechnol 2001. 19:971-974. [DOD] [CrossRef]
- Carpenter MK, Rosler ES, Fisk GJ, Brandenberger R, Ares X, Miura T, Lucero M, Rao MS. Properties of four human embryonic stem cell lines maintained in a feeder-free culture system. Dev Dyn 2004. 229(2):243-258. [DOD] [CrossRef]
- Kroon E, Martinson LA, Kadoya K, Bang AG, Kelly OG, Eliazer S, Young H, Richardson M, Smart NG, Cunningham J, Agulnick AD, D'Amour KA, Carpenter MK, Baetge EE. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat Biotechnol 2008. 26:443-452. [DOD] [CrossRef]
- Liew CG, Shah NN, Briston SJ, Shepherd RM, Khoo CP, Dunne MJ, Moore HD, Cosgrove KE, Andrews PW. PAX4 enhances beta-cell differentiation of human embryonic stem cells. PLoS ONE 2008. 3:e1783. [DOD] [CrossRef]
- International Stem Cell Initiative, Adewumi O, Aflatoonian B, Ahrlund-Richter L, Amit M, Andrews PW, Beighton G, Bello PA, Benvenisty N, Berry LS, et al. Characterization of human embryonic stem cell lines by the International Stem Cell Initiative. Nat Biotechnol 2007. 25(7):803-816. [DOD] [CrossRef]
- Abeyta MJ, Clark AT, Rodriguez RT, Bodnar MS, Pera RA, Firpo MT. Unique gene expression signatures of independently-derived human embryonic stem cell lines. Hum Mol Genet 2004. 13(6):601-608. [DOD] [CrossRef]
- Osafune K, Caron L, Borowiak M, Martinez RJ, Fitz-Gerald CS, Sato Y, Cowan CA, Chien KR, Melton DA. Marked differences in differentiation propensity among human embryonic stem cell lines. Nat Biotechnol 2008. 26(3):313-315. [DOD] [CrossRef]
- Squifflet JP, Moudry K, Sutherland DE. Is HLA matching relevant in pancreas transplantation? Transpl Int 1988. 1(1):26-29. [DOD]
- Taylor CJ, Bolton EM, Pocock S, Sharples LD, Pedersen RA, Bradley JA. Banking on human embryonic stem cells: estimating the number of donor cell lines needed for HLA matching. Lancet 2006. 366:2019-2025. [DOD] [CrossRef]
- Rao MS, Auerbach JM. Estimating human embryonic stem-cell numbers. Lancet 2006. 367:650-650. [DOD] [CrossRef]
- Nakajima F, Tokunaga K, Nakatsuji N. Human leukocyte antigen matching estimations in a hypothetical bank of human embryonic stem cell lines in the Japanese population for use in cell transplantation therapy. Stem Cells 2007. 25:983-985. [DOD] [CrossRef]
This article has been cited by other articles:

|
Genetic analysis of type 1 diabetes: embryonic stem cells as new tools to unlock biological mechanisms in type 1 diabetes
Holmes N, Cooke A
Rev Diabet Stud 2012. 9(4):137-147
|
|

|
Islet-derived stem cells from adult rats participate in the repair of islet damage
Gong J, Zhang G, Tian F, Wang Y
J Mol Histol 2012. Epub
|
|

|
Pancreatic stem/progenitor cells for the treatment of diabetes
Noguchi H
Rev Diabet Stud 2010. 7(2):105-111
|
|

|
Generation of insulin-producing cells from pluripotent stem cells: from the selection of cell sources to the optimization of protocols
Liew CG
Rev Diabet Sud 2010. 7(2):82-92
|
|

|
Islet transplantation in type 1 diabetics using an immunosuppressive protocol based on the anti-LFA-1 antibody efalizumab
Posselt AM, Bellin MD, Tavakol M, Szot GL, Frassetto LA, Masharani U, Kerlan RK, Fong L, Vincenti FG, Hering BJ, Bluestone JA, Stock PG
Am J Transplant 2010. 10(8):1870-1880
|
|
|


)
)
)
)

