Review
| Rev Diabet Stud,
2007,
4(1):13-24 |
DOI 10.1900/RDS.2007.4.13 |
Genes, Diet and Type 2 Diabetes Mellitus: A Review
George V.Z. Dedoussis, Andriana C. Kaliora, Demosthenes B. Panagiotakos
Department of Nutrition and Dietetics, Harokopio University of Athens, 70 El. Venizelou Str., 17671 Kallithea-Athens, Greece.
Address correspondence to: George Dedoussis, e-mail: dedousi@hua.gr
Keywords: diabetes, diet, genes, polymorphism, gene-diet interaction
Abstract
Diabetes mellitus is widely recognized as one of the leading causes of death and disability. While insulin insensitivity is an early phenomenon partly related to obesity, pancreatic β-cell function declines gradually over time even before the onset of clinical hyperglycemia. Several mechanisms have been proposed to be responsible for insulin resistance, including increased non-esterified fatty acids, inflammatory cytokines, adipokines, and mitochondrial dysfunction, as well as glucotoxicity, lipotoxicity, and amyloid formation for β-cell dysfunction. Moreover, the disease has a strong genetic component, although only a handful of genes have been identified so far. Diabetic management includes diet, exercise and combinations of antihyperglycemic drug treatment with lipid-lowering, antihypertensive, and antiplatelet therapy. Since many persons with type 2 diabetes are insulin resistant and overweight, nutrition therapy often begins with lifestyle strategies to reduce energy intake and increase energy expenditure through physical activity. These strategies should be implemented as soon as diabetes or impaired glucose homoeostasis (pre-diabetes) is diagnosed.
Introduction
Diabetes mellitus has been widely recognized to be a fundamental and leading cause of major health issues, in particular of all the cardiovascular diseases. It affects more than 170 million individuals worldwide. There is ample evidence indicating that by 2010, a further growth will occur, mainly caused by dramatic increases in the developing countries of Africa, Asia, and South America [1].
In type 2 diabetes mellitus (T2DM), lifestyle factors, in particular those related to obesity [2], contribute to its development. In industrialized countries, a rapid increase in fat, saturated fatty acid and energy intake accompanies a decrease in physical activity, in the consumption of dietary fiber and of diets with low glycemic index. To date, this development has resulted in a 6% prevalence of T2DM in these populations [3], 4% of whom are obese white adolescents. Furthermore, a prevalence of 25% abnormal glucose tolerance must be reported [4].
Diet and obesity are the main factors affecting the prevalence as well as the development and severity of T2DM. The present paper therefore focuses on dietary and genetic aspects in T2DM in relation to preventing the condition.
Diagnosis of diabetes and obesity
According to WHO recommendations (1999), the diagnosis of diabetes incorporates both fasting and 2-h post-glucose load (75 g) glucose concentrations [5]. Impaired glucose tolerance carries an increased risk of macrovascular disease [6]. Insulin resistance is strongly associated with obesity and physical inactivity. A number of circulating hormones, cytokines, and metabolic fuels, such as non-esterified (free) fatty acids (NEFA), originate in the adipocyte and modulate insulin action. An increased mass of stored triglyceride, especially in visceral or deep subcutaneous adipose depots, leads to large adipocytes that are themselves resistant to the ability of insulin to suppress lipolysis. This results in increased release and circulating levels of NEFA and glycerol, both of which aggravate insulin resistance in skeletal muscle and the liver [7].
Heredity in T2DM
A number of nationwide studies exist on the heritability of diabetes, which show that T2DM, where both genetic and environmental effects play a significant role, tends to run in families. Thus, the risk for T2DM is increased when there is a positive family history of the disease. Approximately 15-25% of the first-degree relatives of patients with T2DM develop either impaired glucose tolerance or diabetes [8]. When the prevalence of diabetes and glucose intolerance was studied, both cross-sectionally and longitudinally, in a cohort of 199 offspring of conjugal diabetic parents, the occurrence of T2DM was estimated to approach 60% by the age of 60 years [9]. The concordance rate for diabetes has been shown to differ among twin pairs. Thus, it ranges between 35% and 58% among monozygotic twins, rising to 88% when impaired glucose tolerance is included [10], and is 17% to 20% among dizygotic twins [11, 12].
Nutritional management in T2DM
Nutrition management is a key component in the long-term health and quality of life of people with T2DM.The general nutrition principles and recommendations are the same for people with diabetes as for those without the condition. However, it is important to set realistic goals that are in tune with the individual's micro- and macronutrient, physical activity, lifestyle and medical needs. The overall goal of diabetes management is to help individuals and their families gain the necessary knowledge, life skills, resources and support needed to achieve optimum health. Nutritional management seeks to improve or maintain the quality of life, physiological health and nutritional status of people with diabetes, by recognizing that their micro- and macronutrient requirements are similar to those of the general population.
The goal of diabetes management is to keep blood glucose levels as close to a normal range as safely possible, while avoiding blood glucose levels that are too high (hyperglycemia) or too low (hypoglycemia) [13]. Attention to food portions and weight management, combined with physical activity, helps improve glycemic control. General guidelines include 50-60% of daily energy requirements derived from carbohydrates, low glycemic index foods, foods containing cereal fiber and a protein intake of least 0.86 g/kg/day. The consumption of added sugars can be up to 10% of daily energy requirements. Also, guidelines recommend the limited intake of total fat, especially saturated fats, with monounsaturated fatty acid (MUFA) used where possible, appropriate use of nutritive and non-nutritive sweeteners, the daily vitamin and mineral requirements of a well balanced diet and individualized physical activity for people with T2DM.
The ongoing efforts to understand the relationships between the genome and diet, termed nutrigenetics and nutrigenomics, have currently modified the definition of T2DM management. Both disciplines aim to unravel diet/genome interactions and identify the optimal diet for individuals, either by clarifying how the genetic makeup of an individual coordinates response to diet [14], or by relating different phenotypes to differences in cellular and/or genetic response (Figure 1).
 |
 |
Figure 1. Simplified model of factors affecting the development of type 2 diabtes. |
|
Candidate and susceptibility genes
The candidate gene approach results in the identification of the gene variants that influence disease. If studies clearly show that a variant has a significant effect on disease-related phenotypes, then this is described as a susceptibility gene. Meta-analyses provide convincing support for the involvement of a gene variant in disease risk. The most compelling evidence generated from a meta-analysis of multiple published studies, results in the determination of the susceptibility genes from among the candidate genes. However, researchers should always consider that if a disease is heterogeneous and polygenic, such as T2DM, studies of single gene variants may not be conclusive and consistent with one another. The contribution of a gene variant to disease and related phenotype is more certain when it is the result of gene-gene interaction studies.
Variants in many candidate genes for T2DM have been extensively studied over the past two decades. Such genes are the KCNJ11, which encodes Kir6.2, the TCF7L2, the Gly972Arg polymorphism in IRS1, the Gly1057Asp polymorphism in IRS2, the Trp64Arg polymorphism in the gene encoding the β3 adrenergic receptor and the -308 G/A promoter variant in TNF (Figure 2). In most instances the initial association was not replicated in subsequent analyses. Even if the adiponectin gene variants do not definitely confer susceptibility for T2DM, they are nevertheless candidate genes.
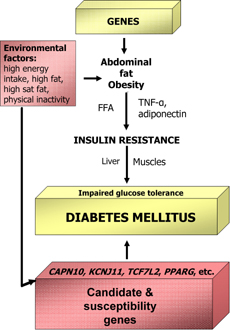 |
|
Figure 2. Interrelation between genes and environmental factors in type 2 diabetes. Genes in combination with environmental factors may lead to obesity, insulin resistance and finally diabetes. Environmental factors may also act in concert together with diabetes candidate and susceptibility genes to trigger the pathogenesis of type 2 diabetes directly. |
|
Calpain-10
The first "common diabetes gene" cloned in this way was CAPN10 in the NIDDM1 region of chromosome 2 [15, 16]. It encodes for calpain-10, a cysteine protease which is ubiquitously expressed [17, 18]. Originally, the G allele of a non-coding single nucleotide polymorphism (UCSNP-43) was associated with T2DM. The risk of developing T2DM, however, is not fully attributable to a single polymorphism of the gene encoding calpain 10, but, as was described in a Mexican-American population, results from the combination of haplotypes created by alleles of three single nucleotide polymorphisms (SNPs): SNP-43, SNP-19, and SNP-63 [16]. All these three SNPs are located within introns and are highly likely to influence gene expression. In later studies, this finding could not be replicated [19, 20], but another SNP (UCSNP-44) was found to be associated with T2DM [21, 22]. Genetic variants in the gene encoding calpain-10 might affect insulin sensitivity, [23] or insulin secretion, [24] or the relation between the two [25].
Kir6.2
Pancreatic β-cell ATP-sensitive K+ channels (KATP channels) are crucial for the regulation of insulin secretion by coupling cell metabolism to membrane electrical activity. The pancreatic β-cell KATP channel comprises two subunits, the inwardly rectifying potassium channel, Kir6.2 (encoded by KCNJ11), and the sulfonylurea receptor, SUR1 (encoded by ABCC8) [26]. The ABCC8 and KCNJ11 genes are located on chromosome 11p15.1 and several gene variants of both genes have been associated with disorders of insulin secretion and T2DM [27, 28]. The strongest statistical association with T2DM in KCNJ11 is with SNPs 74 (3p+215), 76 (A190) and 77 (E23K). However, the evidence suggests a predominant role for the amino acid variant E23K in T2DM susceptibility [28].
TCF7L2
Grant et al. [29] have identified TCF7L2 as a novel susceptibility gene for T2DM and reported an association with the microsatellite marker DG10S478 within intron 3. The associated tetranucleotid repeat was shown to have six alleles, namely alleles 0, 4, 8, 12, 16 and 20. The combined non-zero alleles of DG10S478 (referred to as X) were associated with an increased risk in three independent populations (Danish, Icelandic and US). The composite at-risk allele X was almost perfectly correlated with the T allele of the SNP rs12255372, while the G allele was linked to allele 0. Consequently, the rs12255372 T allele showed an association with T2DM as well [29]. This association was detected in the course of positional cloning efforts in a region of chromosome 10q previously linked to diabetes in Icelandic pedigrees [30] and confirmed in case-control analyses of Danish and U.S. cohorts. Quite recently, the hypothesis that TCF7L2 is a genuine T2DM susceptibility gene was confirmed [31]. Unraveling the mechanisms whereby changes in the function or regulation of this transcription factor lead to loss of β-cell performance and/or insulin sensitivity is likely to provide crucial new insights into disease pathogenesis.
Other genes
Adipose tissue-derived cytokines, TNF-α and IL-6, could be involved in the development of diabetes through several mechanisms. Elevated levels of these cytokines have been linked to the risk for diabetes [32-34]. Genes encoding proteins critical in pancreatic β-cell function are particularly good candidate genes for T2DM. Studies of maturity-onset diabetes in the young in humans [35] and knockout mice [36] have shown that mutations in the transcription factors required for the development, differentiation, and maintenance of the pancreatic β-cells can cause diabetes. EIF4A2, the gene encoding eukaryotic translation initiation factor 4α2, an ubiquitous RNA helicase involved in protein translation initiation, is down-regulated in rat β-INS832/13 cells exposed to high-glucose concentrations. Thus, EIF4A2 may contribute to the regulation of insulin production in response to glucose. Cheyssac et al. [37] recently reported that a SNP in this gene was significantly associated with T2DM diagnosed 45 years ago and with the age of onset in French families.
Hepatocyte nuclear factor (HNF)-4α is a transcription factor known as a key molecule in β-cell development and function. In a previously performed genome-wide scan of Japanese type 2 diabetic sibpairs, researchers observed linkage of T2DM to chromosome 20q12-q13, a region in which the HNF4A gene is located [38]. Recent studies report associations between T2DM and polymorphisms in the P2 promoter region specific to β-cells [39-41].
The ubiquitously expressed protein tyrosine phosphatase-1B (PTP1B), encoded by the PTPN1 gene, catalyzes the dephosphorylation of tyrosine residues from the insulin receptor kinase activation segment [42] and IRS1 [43] resulting in the down-regulation of insulin signaling. PTP1B also inhibits leptin signaling through the dephosphorylation of JAK2 and STAT3 [44, 45]. The disruption of the PTPN1 gene in mice results in increased insulin sensitivity, and in resistance to diet-induced obesity [46], as well as enabling normalization of blood glucose levels [47]. Several studies have investigated the association of T2DM with genetic variants of PTPN1. When analyzing the PTPN1 gene locus, Bento et al. [48] found convincing associations between multiple SNPs and T2DM in two independent Caucasian American case-control samples. Recently published data indicate that PTPN1 variants might modify the lipid profile, thereby influencing susceptibility to the metabolic syndrome [49].
PPARG as a T2DM susceptibility gene
PPARγ is encoded by PPARG, and is a transcription factor that is activated by certain fatty acids, prostanoids, and thiazolidinediones [50, 51]. PPARγ1 is expressed in most tissues, while PPARγ2 is specific to adipose tissue, where it regulates adipogenic differentiation [52]. The high risk proline allele of the Pro12Ala PPARγ polymorphism has a prevalence of 75% in white people. Two meta-analyses and a large prospective analysis have shown a risk reduction of 21-27% for the alanine allele [53-55]. The alanine genotype presumably results in greater insulin sensitivity [56-58]. The proline variant has lower transcriptional activity and heterozygous PPARγ knockout mice are more insulin resistant. Since PPARγ2 is exclusively expressed in adipose tissue, a primary mechanism in this tissue with a secondary effect on hepatic insulin sensitivity and insulin clearance can be invoked [59]. C1431T silent substitution in the 6th exon of PPARG is the polymorphism that modulates the effect of Pro12Ala on susceptibility to T2DM [60]. The PPARG Pro12Ala polymorphism has been shown to be protective against mild fasting hyperglycemia and T2DM during a 6-year follow-up in subjects who were normoglycemic at baseline [61]. The PPARG exon 6 C1431T SNP was also associated with a lower risk.
A significant interaction has also been described for the PPARα Leu162Val polymorphism and n-6 polyunsaturated fatty acid (PUFA) intake. In persons with the less common V162 allele, increased n-6 PUFA intake is associated with a marked reduction in triacylglycerol concentration, whereas this association is not observed in L162 carriers [62].
Adiponectin
Adiponectin levels correlate negatively with glucose, insulin and triglyceride levels as well as the body mass index (BMI), while there is a positive correlation with high-density lipoproteins (HDL), cholesterol levels and insulin-stimulated glucose disposal [63, 64]. Furthermore, high plasma adiponectin concentration protects against T2DM [65]. The adiponectin/ACDC gene is located on chromosome 3q27 at a locus linked to T2DM and also to several phenotypes related to features of the metabolic syndrome [66, 67]. SNPs of APM1, the gene encoding adiponectin, were associated with the development of hyperglycemia [68]. Both case-control and prospective studies in general populations have shown that inherited variations at the ACDC locus modulate adiponectin levels and insulin sensitivity and are part of the genetic background of T2DM [69-72]. Together with animal studies, genetic epidemiology approaches suggest a causative contribution on the part of adiponectin signaling for obesity-associated insulin resistance and T2DM.
Two putative adiponectin receptors, adiponectin receptor 1 (AdipoR1) and adiponectin receptor 1 (AdipoR2), have been cloned [73]. Both receptors are significantly expressed in liver, muscle, and adipose tissue. Their expression levels in muscle correlate with distinct metabolic parameters, particularly with first-phase insulin secretion for AdipoR1 [74], but their relation with insulin sensitivity is not completely established. Expression of both receptors is lower in normal glucose tolerant subjects with a family history of diabetes [75]. Recently, the absence of contributions from ADIPOR1 SNPs in the genetic risk for T2DM was reported [76].
Diet and gene-diet interactions
Nutrition recommendations for the general population are also appropriate for persons with T2DM. Because many persons with T2DM are overweight and insulin resistant, medical nutrition therapy should emphasize lifestyle changes that result in reduced energy intake and increased energy expenditure through physical activity. Many people with diabetes are also diagnosed with dyslipidemia and hypertension, making reductions in dietary intake of saturated fat, cholesterol, and sodium desirable.
Therefore, the emphasis of nutrition therapy for T2DM is given to lifestyle strategies to reduce glycemia, dyslipidemia and blood pressure. These strategies should be implemented as soon as the diagnosis of diabetes is made.
High-fat feeding has been presumed to be a cause of obesity and insulin resistance for at least twenty years [77]. However, the concept that "oils ain't oils" [78] slowly started emerging in the latter half of the eighties and it is now well established that the fatty acid profile of a dietary fat has far-reaching differential regulatory consequences in the human body. High saturated fat content of the membrane makes for rigid, unresponsive membranes, whereas increased desaturation makes for improved membrane fluidity and responsiveness [79]. Diets containing tallow (predominantly saturated fat), olive oil (mainly mono-unsaturated), sunflower oil (largely omega-6) and fish oil (omega-3-rich) are commonly administered to animals when investigating the effect of dietary fat on insulin resistance and obesity. In animal studies, an impressive body of evidence has established the connection between dietary lipids, membrane lipid profiles and insulin resistance [80-82]. A pioneering study performed by Storlien and coworkers in 1987 [78] showed that only by replacing sunflower oil (omega(n)-6) with fish oil (n-3) in rats fed on a high sucrose and high fat diet, was the development of insulin resistance attenuated. In humans, the obesity and T2DM-prone Pima population proved to have 40% lower n-3 levels in their muscle membrane lipids than Australians [83, 84]. Kopecky and coworkers have shown that impairment of the mitochondrial uncoupling protein (UCP) system can lead to obesity and thus to insulin resistance. UCP synthesis is regulated by the PPARγ transcription factor family, which, in turn, can be regulated by fatty acids [85]. Recent studies have supported the concept that polyunsaturated fatty acids can act as ligands of PPARγ [86, 87] or can modulate its expression, thus increasing GLUT4 transcription [88, 89] and synthesis and improving insulin resistance.
Certain polyunsaturated fatty acids, but not saturated fatty acids, can be converted to eicosanoids by cyclooxygenase. For example, AA (omega-6) can be changed to the highly inflammatory prostaglandins of the -2 series, whereas EPA (omega-3) can be converted to the anti-inflammatory prostaglandins of the -3 series. DHA (omega-3) cannot be converted to a prostaglandin, but retroconversion to EPA, and hence formation of series -3 prostaglandins, is possible [90]. Although eicosanoids are implicated in the regulation of GLUT4 trafficking and insulin-stimulated glucose transport [91], Nugent et al. report a cyclooxygenase-independent stimulatory effect of AA on glucose uptake [92].
An interaction between the intake of MUFA, BMI and the Pro12Ala polymorphism of the PPARγ2 gene has recently been reported [93]. It is suggested that an interaction exists between Pro12Ala polymorphism of PPARγ2 and dietary MUFA, which means that obese people with the Ala-12 allele have higher homeostasis model assessment (HOMA) insulin resistance (IR) values, especially if MUFA intake is low [94]. Another gene implicated in gene-diet interaction in diabetes is Rad. It is a prototypic member of the RGK family of Ras-related GTPases, which also includes Gem/Kir, Rem, and Rem2 [95, 96]. Rad was originally found to be overexpressed in the skeletal muscle of a patient with T2DM using subtraction cloning [95]. Ilany et al. have generated transgenic (tg) mice that overexpress Rad in muscle [97]. Rad-Tg mice exhibit normal growth and development, as well as normal glucose homeostasis and insulin sensitivity, as measured by fasting and fed glucose and insulin levels and glucose and insulin tolerance testing. However, when fed a high-fat diet, Rad-Tg mice became more insulin resistant and glucose intolerant than normal mice on the same diet. The combination of high-fat diet and Rad overexpression caused more severely diminished insulin-stimulated glucose uptake than high-fat diet alone, even though Rad overexpression alone did not change glucose transport. Thus, Rad overexpression interacts with a high-fat diet to worsen insulin resistance in muscle. This finding was consistent with clinical studies suggesting that Rad may interact with obesity in increasing diabetes risk [98] and is an example of how a genetic factor (Rad overexpression) can act together with an environmental factor (high-fat diet and obesity) to alter glucose homeostasis.
Perilipin, which is encoded by PLIN, is the predominant protein associated with lipid storage droplets in adipocytes and one of the critical regulators implicated in lipid mobilization [99, 100]. The connection between perilipin, body fat, and insulin resistance has been demonstrated in knockout mouse models, which displayed reduced body fat and, paradoxically, an increased risk of glucose intolerance and peripheral insulin resistance [100]. Dietary factors play a role in the relationship between perilipin and body weight. PLIN polymorphism modulates weight loss in response to a low-energy diet [101]. Genetic variation at the PLIN locus modulates the effects of habitual dietary fat and carbohydrate consumption on insulin resistance in a large sample of an Asian female general population. A statistically significant gene-diet interaction was found between intake of fat and carbohydrate and polymorphisms in PLIN (11482GA and 14995AT) [102].
SCARB1 genetic variability plays a significant role in lipoprotein metabolism in humans [103, 104]. Recent findings show that the presence of the A allele at the SCARB1 exon 1 polymorphism is associated with a statistically significant increase in insulin sensitivity after the consumption of a MUFA-rich diet compared to the effect on G/G individuals [105]. The fatty acid (FA)-binding protein 2 (FABP2) gene codes for intestinal FABP (IFABP), which is a member of a family of small (14-15-kDa) intracellular lipid-binding proteins. The gene located at 4q28-q31 has the conserved 4 exons and 3 introns that are characteristic of this family of genes [106, 107]. IFABP is crucial for fat absorption and transport: the uptake and trafficking of saturated and unsaturated long-chain fatty acids (LCFAs), the targeting of free fatty acids (FFAs) toward different metabolic pathways, protection of the cytosol from the cytotoxic effects of FFAs and the modulation of the enzyme additive involved in lipid metabolism [108, 109]. Besides FFAs, IFABP may bind other ligands such as phenolic antioxidants. It is abundant in the enterocyte, representing 2-3% of the cytoplasmic mass of those cells [110]. It has been found that the expression of IFABP mRNA is under dietary control [111]. Subjects with the Thr54 allele had higher FFA concentrations than did those who were homozygous for the Ala54 allele when consuming a saturated fatty acid (SFA)-rich diet [112]. Considering the hypothesis proposed by Baier et al. [113], this suggests a plausible mechanism for the FABP2 Ala54/Thr54 polymorphism-diet interaction for the determination of insulin sensitivity. Thus, the presence of the FABP2 gene Ala54/Thr54 polymorphism impairs peripheral insulin sensitivity when the carriers consume an SFA diet.
Conclusions
The importance of preventing diabetes in high-risk individuals is highlighted by the substantial and worldwide increase in the prevalence of diabetes in recent years. Genetic susceptibility appears to play a powerful role in the occurrence of T2DM in certain populations. However, given that population gene pools shift quite slowly, the current epidemic most likely reflects marked changes in lifestyle. Lifestyle changes that are characterized by decreased physical activity and increased energy consumption have combined to promote obesity, a strong risk factor for diabetes that is influenced itself by both genes and behavior (Figure 2). Despite the difficulty in maintaining a reduced body weight long-term, several studies have demonstrated that there is potential for moderate sustained weight loss in order to substantially reduce the risk for T2DM. Reduced intake of total fat, particularly saturated fat, may reduce risk for diabetes. Increased diabetes incidence is reported with increased intake of dietary fat, independent of total calories, although this effect is not demonstrated in all studies. The importance of a balanced ratio of omega-6 to omega-3 intake, as in the ancient Paleolithic diet, was recently tested in the Lyon Heart Study [114]. This study was a prospective, randomized, single-blinded secondary prevention trial, which compared the effects of a modified Cretan diet, enriched with alpha-linolenic acid (ALA; ratio of omega-6/3, 4:1), low in saturated fat, very low in trans fat and high in vitamin C and E, to that of a Step I American Heart Association Diet in the secondary prevention of coronary events and death. It would appear to be prudent to return to the hunter-gatherer diet of our ancestors or the suggested Mediterranean (Crete) type diet [114, 115] that contains less saturated and trans-fatty acids and more polyunsaturated fatty acids with an improved omega-6/omega-3 ratio [115]. Studies in healthy subjects suggest that diets based on olive oil, as opposed to diets rich in SFA or PUFA, have beneficial effects on atherogenic factors linked to inflammation of the vascular wall, such as monocyte chemotaxis and adhesion to endothelial cells and expression of adhesion molecules [116, 117]. In this regard, treatment of endothelial cells with oleic acid protected them against cytokine-induced adhesion molecule overexpression [118].
Moderate reductions in dietary sodium intake (2400 mg/day) decreased blood pressure by 5 mm Hg systolic and 2 mm Hg diastolic in hypertensive patients and by 3 mm Hg systolic and 1 mm Hg diastolic in normotensive patients [119]. Although there is a wide variation in blood pressure responses, the lower the sodium intake, the greater the lowering of blood pressure [120]. Responses to sodium reduction may be greater in subjects who are "salt sensitive", a characteristic of many individuals with diabetes [121].
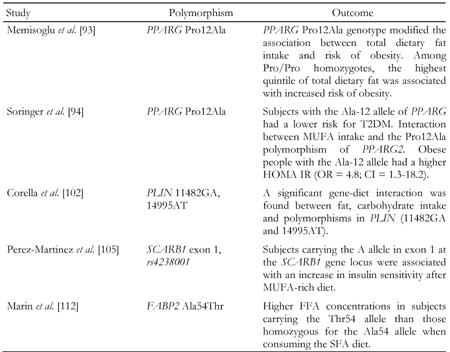
The so-called prudent dietary pattern, characterized by a high intake of vegetables, legumes, fruit, and whole grains and a low intake of red meat, processed meat, high-fat dairy products, and refined grains is increasingly favored for a beneficial effect on a number of health outcomes, including diabetes treatment and prevention. The prudent diet does not necessarily have to be a low-fat one. A variety of vegetable foods rich in MUFA that are good sources of antioxidants, such as high-oleic acid oils and nuts, can be incorporated into this dietary pattern to increase palatability and compliance, with a high chance of health benefits. Together with a healthy diet, unraveling the plethora of diabetes-predisposing genes and elucidation of their interactions with the dietary products (Table 1) will probably reduce the incidence of T2DM and improve the outcome of the disease.
Table
1.
Summary of major results on gene-diet interactions in the development of T2DM |
|
 |
|
Legend:
MUFA: monounsaturated fatty acid. HOMA: homeostasis model assessment. IR: insulin resistance. SFA: saturated fatty acid. FFA: free fatty acids. T2DM: type 2 diabetes mellitus. OR: odds ratio. CI: confidence interval. |
|
References
- Zimmet P, Alberti KG, Shaw J. Global and societal implications of the diabetes epidemic. Nature 2001. 414:782-787. [DOD] [CrossRef]
- Caballero B. A nutrition paradox - underweight and obesity in developing countries. N Engl J Med 2005. 352:1514-1516. [DOD] [CrossRef]
- King H, Aubert RE, Herman WH. Global burden of diabetes, 1995-2025: prevalence, numerical estimates, and projections. Diabetes Care 1998. 21:1414-1431. [DOD] [CrossRef]
- Sinha R, Fisch G, Teague B, Tamborlane WV, Banyas B, Allen K, Savoye M, Rieger V, Taksali S, Barbetta G, et al. Prevalence of impaired glucose tolerance among children and adolescents with marked obesity. N Engl J Med 2002. 346:802-810. [DOD] [CrossRef]
- World Health Organization Expert Committee. Definition, diagnosis and classification of diabetes mellitus and its complications. Report of a WHO consultation, part 1: diagnosis and classification of diabetes mellitus. WHO, Geneva, 1999. [DOD]
- DECODE Study Group. Glucose tolerance and mortality:comparison of WHO and American Diabetes Association diagnostic criteria. The DECODE study group. European Diabetes Epidemiology Group. Diabetes Epidemiology: Collaborative analysis of diagnostic criteria in Europe. Lancet 1999. 354:617-621. [DOD] [CrossRef]
- Boden G. Role of fatty acids in the pathogenesis of insulin resistance and NIDDM. Diabetes 1997. 46:3-10. [DOD] [CrossRef]
- Pierce M, Keen H, Bradley C. Risk of diabetes in offspring of parents with non-insulin-dependent diabetes. Diabet Med 1995. 12:6-13. [DOD]
- Tattersal RB, Fajans SS. Prevalence of diabetes and glucose intolerance in 199 offspring of thirty-seven conjugal diabetic parents. Diabetes 1975. 24:452-462. [DOD] [CrossRef]
- Henkin L, Bergman RN, Bowden DW, Ellsworth DL, Haffner SM, Langefeld CD, Mitchell BD, Norris JM, Rewers M, Saad MF, et al. Genetic epidemiology of insulin resistance and visceral adiposity. The IRAS Family Study design and methods. Ann Epidemiol 2003. 13:211-217. [DOD] [CrossRef]
- Kaprio J, Tuomilehto J, Koskenvuo M, Romanov K, Reunanen A, Eriksson J, Stengard J, Kesaniemi YA. Concordance for type 1 (insulin-dependent) and type 2 (non-insulin-dependent) diabetes mellitus in a population-based cohort of twins in Finland. Diabetologia 1992. 35:1060-1067. [DOD] [CrossRef]
- Newman B, Selby JV, King MC, Slemenda C, Fabsitz R, Friedman GD. Concordance for type 2 (non-insulin-dependent) diabetes mellitus in male twins. Diabetologia 1987. 30:763-768. [DOD] [CrossRef]
- Moran M. The evolution of the nutritional management of diabetes. Proc Nutr Soc 2004. 63:615-620. [DOD] [CrossRef]
- Ordovas JM, Mooser V. Nutrigenomics and nutrigenetics. Curr Opin Lipidol 2004. 15:101-108. [DOD] [CrossRef]
- Cox NJ. Challenges in identifying genetic variation affecting susceptibility to type 2 diabetes: examples from studies of the calpain-10 gene. Hum Mol Genet 2001. 10:2301-2305. [DOD] [CrossRef]
- Horikawa Y, Oda N, Cox NJ, Li X, Orho-Melander M, Hara M, Hinokio Y, Lindner TH, Mashima H, Schwarz PE, et al. Genetic variation in the gene encoding calpain-10 is associated with type 2 diabetes mellitus. Nat Genet 2000. 26:163-175. [DOD] [CrossRef]
- Ma H, Fukiage C, Kim YH, Duncan MK, Reed NA, Shih M, Azuma M, Shearer TR. Characterization and expression of calpain 10. A novel ubiquitous calpain with nuclear localization. J Biol Chem 2001. 276:28525-28531. [DOD] [CrossRef]
- Suzuki K, Hata S, Kawabata Y, Sorimachi H. Structure, activation, and biology of calpain. Diabetes 2004. 53:S12-S18. [DOD] [CrossRef]
- Rasmussen SK, Urhammer SA, Berglund L, Jensen JN, Hansen L, Echwald SM, Borch-Johnsen K, Horikawa Y, Mashima H, Lithell H, et al. Variants within the calpain-10 gene on chromosome 2q37 (NIDDM1) and relationships to type 2 diabetes, insulin resistance, and impaired acute insulin secretion among Scandinavian Caucasians. Diabetes 2002. 51:3561-3567. [DOD] [CrossRef]
- Horikawa Y, Oda N, Yu L, Imamura S, Fujiwara K, Makino M, Seino Y, Itoh M, Takeda J. Genetic variations in calpain-10 gene are not a major factor in the occurrence of type 2 diabetes in Japanese. J Clin Endocrinol Metab 2003. 88:244-247. [DOD] [CrossRef]
- Song Y, Niu T, Manson JE, Kwiatkowski DJ, Liu S. Are variants in the CAPN10 gene related to risk of type 2 diabetes? A quantitative assessment of population and family-based association studies. Am J Hum Genet 2004. 74:208-222. [DOD] [CrossRef]
- Weedon MN, Schwarz PE, Horikawa Y, Iwasaki N, Illig T, Holle R, Rathmann W, Selisko T, Schulze J, Owen KR, et al. Meta-analysis and a large association study confirm a role for calpain-10 variation in type 2 diabetes susceptibility. Am J Hum Genet 2003. 73:1208-1212. [DOD] [CrossRef]
- Baier LJ, Permana PA, Yang X, Pratley RE, Hanson RL, Shen GQ, Mott D, Knowler WC, Cox NJ, Horikawa Y, et al. A calpain-10 gene polymorphism is associated with reduced muscle mRNA levels and insulin resistance. J Clin Invest 2000. 106:R69-R73. [DOD]
- Sreenan SK, Zhou YP, Otani K, Hansen PA, Currie KP, Pan CY, Lee JP, Ostrega DM, Pugh W, Horikawa Y, et al. Calpains play a role in insulin secretion and action. Diabetes 2001. 50(9):2013-2020. [DOD] [CrossRef]
- Tripathy D, Eriksson KF, Orho-Melander M, Fredriksson J, Ahlqvist G, Groop L. Parallel manifestation of insulin resistance and beta cell decompensation is compatible with a common defect in Type 2 diabetes. Diabetologia 2004. 47:782-793. [DOD] [CrossRef]
- Seino S. ATP-sensitive potassium channels: a model of heteromultimeric potassium channel/receptor assemblies. Ann Rev Physiol 1999. 61:337-362. [DOD] [CrossRef]
- Barroso I, Luan J, Middelberg RP, Harding AH, Franks PW, Jakes RW, Clayton D, Schafer AJ, O'Rahilly S, Wareham NJ. Candidate gene association study in type 2 diabetes indicates a role for genes involved in beta-cell function as well as insulin action. PLoS Biol 2003. 1(1):E20. [DOD] [CrossRef]
- Hani EH, Boutin P, Durand E, Inoue H, Permutt MA, Velho G, Froguel P. Missense mutations in the pancreatic islet beta cell inwardly rectifying KC channel gene (KIR6.2/BIR): a meta-analysis suggests a role in the polygenic basis of type II diabetes mellitus in Caucasians. Diabetologia 1998. 41(12):1511-1515. [DOD] [CrossRef]
- Grant SF, Thorleifsson G, Reynisdottir I, Benediktsson R, Manolescu A, Sainz J, Helgason A, Stefansson H, Emilsson V, Helgadottir A, et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet 2006. 38:320-323. [DOD] [CrossRef]
- Reynisdottir I, Thorleifsson G, Benediktsson R, Sigurdsson G, Emilsson V, Einarsdottir AS, Hjorleifdottir EE, Orlygsdottir GT, Bjornsdottir GT, Saemundsdottir J, et al. Localization of a susceptibility gene for type 2 diabetes to chromosome 5q34-q35.2. Am J Hum Genet 2003. 73:323-335. [DOD] [CrossRef]
- Groves CJ, Zeggini E, Minton J, Frayling TM, Weedon MN, Rayner NW, Hitman GA, Walker M, Wiltshire S, Hattersley AT, McCarthy MI. Association analysis of 6,736 UK subjects provides replication and confirms TCF7L2 as a type 2 diabetes susceptibility gene with a substantial effect on individual risk. Diabetes 2006. 55:2640-2644. [DOD] [CrossRef]
- Pickup JC, Mattock MB, Chusney GD, Burt D. NIDDM as a disease of the innate immune system: association of acute-phase reactants and interleukin-6 with metabolic syndrome X. Diabetologia 1997. 40(11):1286-1292. [DOD] [CrossRef]
- Nilsson J, Jovinge S, Niemann A, Reneland R, Lithell H. Relation between plasma tumor necrosis factor-alpha and insulin sensitivity in elderly men with non-insulin-dependent diabetes mellitus. Arterioscler Thromb Vasc Biol 1998. 18:1199-1202. [DOD]
- Pradhan AD, Manson JE, Rifai N, Buring JE, Ridker PM. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA 2001. 286:327-334. [DOD] [CrossRef]
- Pozzilli P, Di Mario U. Autoimmune diabetes not requiring insulin at diagnosis (latent autoimmune diabetes of the adult): definition, characterization, and potential prevention. Diabetes Care 2001. 24:1460-1467. [DOD] [CrossRef]
- Habener JF, Stoffers DA. A newly discovered role of transcription factors involved in pancreas development and the pathogenesis of diabetes mellitus. Proc Assoc Am Physicians 1998. 110:12-21. [DOD]
- Cheyssac C, Dina C, Lepretre F, Vasseur-Delannoy V, Dechaume A, Lobbens S, Balkau B, Ruiz J, Charpentier G, Pattou F, et al. EIF4A2 is a positional candidate gene at the 3q27 locus linked to type 2 diabetes in French families. Diabetes 2006. 55:1171-1176. [DOD] [CrossRef]
- Mori Y, Otabe S, Dina C, Yasuda K, Populaire C, Lecoeur C, Vatin V, Durand E, Hara K, Okada T, et al. Genomewide search for type 2 diabetes in Japanese affected sib-pairs confirms susceptibility genes on 3q, 15q, and 20q and identifies two new candidate loci on 7p and 11p. Diabetes 2002. 51:1247-1255. [DOD] [CrossRef]
- Damcott CM, Hoppman N, Ott SH, Reinhart LJ, Wang J, Pollin TI, O'Connell JR, Mitchell BD, Shuldiner AR. Polymorphisms in both promoters of hepatocyte nuclear factor 4a are associated with type 2 diabetes in the Amish. Diabetes 2004. 53:3337-3341. [DOD] [CrossRef]
- Silander K, Mohlke KL, Scott LJ, Peck EC, Hollstein P, Skol AD, Jackson AU, Deloukas P, Hunt S, Stavrides G, et al. Genetic variation near the hepatocyte nuclear factor-4 α gene predicts susceptibility to type 2 diabetes. Diabetes 2004. 53:1141-1149. [DOD] [CrossRef]
- Hara K, Horikoshi M, Kitazato H, Ito C, Noda M, Ohashi J, Froguel P, Tokunaga K, Tobe K, Nagai R, Kadowaki T. Hepatocyte nuclear factor-4alpha P2 promoter haplotypes are associated with type 2 diabetes in the Japanese population. Diabetes 2006. 55:1260-1264. [DOD] [CrossRef]
- Seely BL, Staubs PA, Reichart DR, Berhanu P, Milarski KL, Saltiel AR, Kusari J, Olefsky JM. Protein tyrosine phosphatase 1B interacts with the activated insulin receptor. Diabetes 1996. 45:1379-1385. [DOD] [CrossRef]
- Goldstein BJ, Bittner-Kowalczyk A, White MF, Harbeck M. Tyrosine dephosphorylation and deactivation of insulin receptor substrate-1 by protein-tyrosine phosphatase 1B. Possible facilitation by the formation of a ternary complex with the Grb2 adaptor protein. J Biol Chem 2000. 275:4283-4289. [DOD] [CrossRef]
- Zabolotny JM, Bence-Hanulec KK, Stricker-Krongrad A, Haj F, Wang Y, Minokoshi Y, Kim YB, Elmquist JK, Tartaglia LA, Kahn BB, Neel BG. PTP1B regulates leptin signal transduction in vivo. Dev Cell 2002. 2:489-495. [DOD] [CrossRef]
- Cheng A, Uetani N, Simoncic PD, Chaubey VP, Lee-Loy A, McGlade CJ, Kennedy BP, Tremblay ML. Attenuation of leptin action and regulation of obesity by protein tyrosine phosphatase 1B. Dev Cell 2002. 2:497-503. [DOD] [CrossRef]
- Elchebly M, Payette P, Michaliszyn E, Cromlish W, Collins S, Loy AL, Normandin D, Cheng A, Himms-Hagen J, Chan CC, et al. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science 1999. 283:1544-1548. [DOD] [CrossRef]
- Klaman LD, Boss O, Peroni OD, Kim JK, Martino JL, Zabolotny JM, Moghal N, Lubkin M, Kim YB, Sharpe AH, et al. Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Mol Cell Biol 2000. 20:5479-5489. [DOD] [CrossRef]
- Bento JL, Palmer ND, Mychaleckyj JC, Lange LA, Langefeld CD, Rich SS, Freedman BI, Bowden DW. Association of protein tyrosine phosphatase 1B gene polymorphisms with type 2 diabetes. Diabetes 2004. 53:3007-3012. [DOD] [CrossRef]
- Cheyssac C, Lecoeur C, Dechaume A, Bibi A, Charpentier G, Balkau B, Marre M, Froguel P, Gibson F, Vaxillaire M. Analysis of common PTPN1 gene variants in type 2 diabetes, obesity and associated phenotypes in the French population. BMC Med Genet 2006. 7:44. [DOD] [CrossRef]
- Olefsky JM. Treatment of insulin resistance with peroxisome proliferator-activated receptor gamma agonists. J Clin Invest 2000. 106:467-472. [DOD]
- Schoonjans K, Auwerx J. Thiazolidinediones: an update. Lancet 2000. 355:1008-1010. [DOD] [CrossRef]
- Auwerx J. PPARgamma, the ultimate thrifty gene. Diabetologia 1999. 42:1033-1049. [DOD] [CrossRef]
- Parikh H, Groop L. Candidate genes for type 2 diabetes. Rev Endocr Metab Disord 2004. 5:151-176. [DOD] [CrossRef]
- Lohmueller KE, Pearce CL, Pike M, Lander ES, Hirschhorn JN. Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nat Genet 2003. 33:177-182. [DOD] [CrossRef]
- Memisoglu A, Hu FB, Hankinson SE, Manson JE, De Vivo I, Willett WC, Hunter DJ. Prospective study of the association between the proline to alanine codon 12 polymorphism in the PPARgamma gene and type 2 diabetes. Diabetes Care 2003. 26:2915-2917. [DOD] [CrossRef]
- Deeb SS, Fajas L, Nemoto M, Pihlajamaki J, Mykkanen L, Kuusisto J, Laakso M, Fujimoto W, Auwerx J. A Pro12Ala substitution in PPARgamma2 associated with decreased receptor activity, lower body mass index and improved insulin sensitivity. Nat Genet 1998. 20(3):284-287. [DOD] [CrossRef]
- Ek J, Andersen G, Urhammer SA, Hansen L, Carstensen B, Borch-Johnsen K, Drivsholm T, Berglund L, Hansen T, Lithell H, Pedersen O. Studies of the Pro12Ala polymorphism of the peroxisome proliferator-activated receptor-γ2 (PPAR-gamma2) gene in relation to insulin sensitivity among glucose tolerant Caucasians. Diabetologia 2001. 44:1170-1176. [DOD] [CrossRef]
- Muller YL, Bogardus C, Beamer BA, Shuldiner AR, Baier LJ. A functional variant in the peroxisome proliferator-activated receptor gamma2 promoter is associated with predictors of obesity and type 2 diabetes in Pima Indians. Diabetes 2003. 52:1864-1871. [DOD] [CrossRef]
- Stumvoll M, Haring H. The Pro12Ala polymorphism in the peroxisome proliferator-activated receptor gamma. Diabetes 2002. 51:2341-2347. [DOD] [CrossRef]
- Doney AS, Fischer B, Cecil JE, Boylan K, McGuigan FE, Ralston SH, Morris AD, Palmer CN. Association of the Pro12Ala and C1431 variants of PPARG and their haplotypes with susceptibility to type 2 diabetes. Diabetologia 2004. 47(3):555-558. [DOD] [CrossRef]
- Jaziri R, Lobbens S, Aubert R, Pean F, Lahmidi S, Vaxillaire M, Porchay I, Bellili N, Tichet J, Balkau B, et al. The PPARG Pro12Ala polymorphism is associated with a decreased risk of developing hyperglycemia over 6 years and combines with the effect of the APM1 G-11391A single nucleotide polymorphism: the Data From an Epidemiological Study on the Insulin Resistance Syndrome (DESIR) study. Diabetes 2006. 55:1157-1162. [DOD]
- Dwyer JH, Allayee H, Dwyer KM, Fan J, Wu H, Mar R, Lusis AJ, Mehrabian M. Arachidonate 5-lipoxygenase promoter genotype, dietary arachidonic acid, and atherosclerosis. N Engl J Med 2004. 350:29-37. [DOD] [CrossRef]
- Weyer C, Funahashi T, Tanaka S, Hotta K, Matsuzawa Y, Pratley RE, Tataranni PA. Hypoadiponectinemia in obesity and type 2 diabetes: close association with insulin resistance and hyperinsulinemia. J Clin Endocrinol Metab 2001. 86:1930-1935. [DOD] [CrossRef]
- Matsubara M, Maruoka S, Katayose S. Decreased plasma adiponectin concentrations in women with dyslipidemia. J Clin Endocrinol Metab 2002. 87:2764-2769. [DOD] [CrossRef]
- Lindsay RS, Funahashi T, Hanson RL, Matsuzawa Y, Tanaka S, Tataranni PA, Knowler WC, Krakoff J. Adiponectin and development of type 2 diabetes in the Pima Indian population. Lancet 2002. 360:57-58. [DOD] [CrossRef]
- Vionnet N, Hani El-H, Dupont S, Gallina S, Francke S, Dotte S, De Matos F, Durand E, Lepretre F, Lecoeur C, et al. Genomewide search for type 2 diabetes-susceptibility genes in French whites: evidence for a novel susceptibility locus for early onset diabetes on chromosome 3q27-qter and independent replication of a type 2-diabetes locus on chromosome 1q21-q24. Am J Hum Genet 2000. 67:1470-1480. [DOD] [CrossRef]
- Kissebah AH, Sonnenberg GE, Myklebust J, Goldstein M, Broman K, James RG, Marks JA, Krakower GR, Jacob HJ, Weber J, et al. Quantitative trait loci on chromosomes 3 and 17 influence phenotypes of the metabolic syndrome. Proc Natl Acad Sci U S A 2000. 97:14478-14483. [DOD] [CrossRef]
- Fumeron F, Aubert R, Siddiq A, Betoulle D, Pean F, Hadjadj S, Tichet J, Wilpart E, Chesnier MC, Balkau B, et al. Adiponectin gene polymorphisms and adiponectin levels are independently associated with the development of hyperglycemia during a 3-year period: the epidemiologic data on the insulin resistance syndrome prospective study. Diabetes 2004. 53:1150-1157. [DOD] [CrossRef]
- Vasseur F, Helbecque N, Dina C, Lobbens S, Delannoy V, Gaget S, Boutin P, Vaxillaire M, Lepretre F, Dupont S, et al. Single nucleotide polymorphism haplotypes in the both proximal promoter and exon 3 of APM1 gene modulate adipocyte-secreted adiponectin hormone levels and contribute to the genetic risk for type 2 diabetes in French Caucasians. Hum Mol Genet 2002. 11:2607-2614. [DOD] [CrossRef]
- Hara K, Boutin P, Mori Y, Tobe K, Dina C, Yasuda K, Yamauchi T, Otabe S, Okada T, Eto K, et al. Genetic variation in the gene encoding adiponectin is associated with an increased risk of type 2 diabetes in the Japanese population. Diabetes 2002. 51:536-540. [DOD] [CrossRef]
- Fumeron F, Aubert R, Siddiq A, Betoulle D, Pean F, Hadjadj S, Tichet J, Wilpart E, Chesnier MC, Balkau B, et al. Adiponectin gene polymorphisms and adiponectin levels are independently associated with the development of hyperglycemia during a 3-year period: the Epidemiologic Data on the Insulin Resistance Syndrome prospective study. Diabetes 2004. 53:1150-1157. [DOD] [CrossRef]
- Vasseur F, Lepretre F, Lacquemant C, Froguel P. The genetics of adiponectin. Curr Diab Rep 2003. 3:151-158. [DOD]
- Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, Sugiyama T, Miyagishi M, Hara K, Tsunoda M, et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 2003. 423:762-769. [DOD] [CrossRef]
- Staiger H, Kaltenbach S, Staiger K, Stefan N, Fritsche A, Guirguis A, Peterfi C, Weisser M, Machicao F, Stumvoll M, Haring HU. Expression of adiponectin receptor mRNA in human skeletal muscle cells is related to in vivo parameters of glucose and lipid metabolism. Diabetes 2004. 53:2195-2201. [DOD] [CrossRef]
- Civitarese AE, Jenkinson CP, Richardson D, Bajaj M, Cusi K, Kashyap S, Berria R, Belfort R, DeFronzo RA, Mandarino LJ, Ravussin E. Adiponectin receptors gene expression and insulin sensitivity in non-diabetic Mexican Americans with or without a family history of type 2 diabetes. Diabetologia 2004. 47:816-820. [DOD] [CrossRef]
- Vaxillaire M, Dechaume A, Vasseur-Delannoy V, Lahmidi S, Vatin V, Lepretre F, Boutin P, Hercberg S, Charpentier G, Dina C, Froguel P. Genetic analysis of ADIPOR1 and ADIPOR2 candidate polymorphisms for type 2 diabetes in the Caucasian population. Diabetes 2006. 55:856-861. [DOD] [CrossRef]
- Storlien LH, James DE, Burleigh KM, Chisholm DJ, Kraegen EW. Fat feeding causes widespread insulin resistance, decreased energy expenditure and obesity in rats. Am J Physiol 1986. 251:E576-E583. [DOD]
- Storlien LH, Kraegen EW, Chisholm DJ, Ford GL, Bruce DG, Pascoe WS. Fish oil prevents insulin resistance induced by high-fat feeding in rats. Science 1987. 237:885-888. [DOD] [CrossRef]
- Okada T, Kawano Y, Sakakibara T, Hazeki O, Ui M. Essential role of phosphatidylinositol-3-kinase in insulin-induced glucose transport and antilipolysis in rat adipocytes. Studies with a selective inhibitor wortmannin. J Biol Chem 1994. 269:3568-3573. [DOD]
- Storlien LH, Jenkins AB, Chisholm DJ, Pascoe WS, Khouri S, Kraegen EW. Influence of dietary fat composition on development of insulin resistance in rats. Relationship to muscle triglyceride and w-3 fatty acids in muscle phospholipids. Diabetes 1991. 40:280-289. [DOD] [CrossRef]
- Borkman M, Storlien LH, Pan DA, Jenkins AB, Chisholm DJ, Campbell LV. The relationship between insulin sensitivity and the fatty acid composition of phospholipids of skeletal muscle. N Eng J Med 1993. 328:328-344. [DOD] [CrossRef]
- Storlien LH, Baur LA, Kriketos AD, Pan DA, Cooney GJ, Jenkins AB, Calvert GD, Campbell LV. Dietary fats and insulin action. Diabetologia 1996. 39:621-631. [DOD]
- Vessby B, Tengblad S, Lithell H. Insulin sensitivity is related to the fatty acid composition of serum lipids and skeletal muscle phospholipids in 70-year-old men. Diabetologia 1994. 37:1044-1050. [DOD]
- Pan DA, Lillioja S, Milner MR, Kriketos AD, Baur LA, Bogardus C, Storlien LH. Skeletal muscle membrane lipid composition is related to adiposity and insulin action. J Clin Invest 1995. 96:2802-2808. [DOD]
- Kopecky J, Flachs P, Bardova K, Brauner P, Prazak T, Sponarova J. Modulation of lipid metabolism by energy status of adipocytes. Ann NY Acad Sci 2002. 967:88-101. [DOD]
- Nugent C, Prins JB, Whitehead JP, Wentworth JM, Chatterjee VK, O'Rahilly S. Arachidonic acid stimulates glucose uptake in 3T3-L1 adipocytes by increasing GLUT1 and GLUT4 levels at the plasma membrane. J Biol Chem 2001. 276:9149-9157. [DOD] [CrossRef]
- Barroso I, Gurnell M, Crowley VE, Agostini M, Schwabe JW, Soos MA, Maslen GL, Williams TD, Lewis H, Schafer AJ, et al. Dominant negative mutations in human PPARgamma associated with severe insulin resistance, diabetes mellitus and hypertension. Nature 1999. 402:880-883. [DOD]
- Kliewer SA, Sundseth SS, Jones SA, Brown PJ, Wisely GB, Koble CS, Devchand P, Wahli W, Willson TM, Lenhard JM, Lehmann JM. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors alpha and gamma. Proc Natl Acad Sci USA 1997. 94:4318-4323. [DOD] [CrossRef]
- Wu Z, Xie Y, Morrison RF, Bucher NL, Farmer SR. PPARgamma induces insulin-dependent glucose transporter GLUT4 in the absence of C/EBPalpha during the conversion of 3T3 fi broblasts into adipocytes. J Clin Invest 1998. 101:22-32. [DOD]
- Atkinson TG, Barker HJ, Meckling-Gill KA. Incorporation of long-chain n-3 fatty acids in tissues and enhanced bone-marrow cellularity with docosahexanoic acid feeding in post-weanling Fischer 344 rats. Lipids 1997. 32:293-302. [DOD] [CrossRef]
- Dransfeld O, Rakatzi I, Sasson S, Gruzman A, Schmitt M, Haussinger D, Eckel J. Eicosanoids participate in the regulation of glucose transport by contribution to a rearrangement of actin cytoskeletal elements. Biochemical Journal 2001. 359:47-54. [DOD] [CrossRef]
- Nugent C, Prins JB, Whitehead JP, Wentworth JM, Chatterjee VK, O'Rahilly S. Arachidonic acid stimulates glucose uptake in 3T3-L1 adipocytes by increasing GLUT1 and GLUT4 levels at the plasma membrane. J Biol Chem 2001. 276:9149-9157. [DOD] [CrossRef]
- Memisoglu A, Hu FB, Hankinson S, Manson J, De Vivo I, Willett WC, Hunter DJ. Interaction between a peroxisome proliferator-activated receptor gene polymorphism and dietary fat intake in relation to body mass. Hum Mol Genet 2003. 12:2923-2929. [DOD] [CrossRef]
- Soriguer F, Morcillo S, Cardona F, Rojo-Martinez G, de la Cruz Almaraz M, Ruiz de Adana Mde L, Olveira G, Tinahones F, Esteva I. Pro12Ala polymorphism of the PPARG2 gene is associated with type 2 diabetes mellitus and peripheral insulin sensitivity in a population with a high intake of oleic acid. J Nutr 2006. 136:2325-2330. [DOD]
- Reynet C, Kahn CR. Rad: a member of the Ras family overexpressed in muscle of type II diabetic humans. Science 1993. 262:1441-1444. [DOD] [CrossRef]
- Maguire J, Santoro T, Jensen P, Siebenlist U, Yewdell J, Kelly K. Gem: an induced, immediate early protein belonging to the Ras family. Science 1994. 265:241-244. [DOD] [CrossRef]
- Ilany J, Bilan PJ, Kapur S, Caldwell JS, Patti ME, Marette A, Kahn CR. Overexpression of Rad in muscle worsens diet-induced insulin resistance and glucose intolerance and lowers plasma triglyceride level. Proc Natl Acad Sci U S A 2006. 103:4481-4486. [DOD]
- Garvey WT, Maianu L, Kennedy A, Wallace P, Ganaway E, Hamacher LL, Yarnall DP, Lenhard JM, Burns DK. Muscle Rad expression and human metabolism: potential role of the novel Ras-related GTPase in energy expenditure and body composition. Diabetes 1997. 46:444-450. [DOD] [CrossRef]
- Martinez-Botas J, Anderson JB, Tessier D, Lapillonne A, Chang BH, Quast MJ, Gorenstein D, Chen KH, Chan L. Absence of perilipin results in leanness and reverses obesity in Lepr (db/db) mice. Nat Genet 2000. 26:474-479. [DOD] [CrossRef]
- Tansey JT, Sztalryd C, Gruia-Gray J, Roush DL, Zee JV, Gavrilova O, Reitman ML, Deng CX, Li C, Kimmel AR, Londos C. PLIN ablation results in a lean mouse with aberrant adipocyte lipolysis, enhanced leptin production, and resistance to diet-induced obesity. Proc Natl Acad Sci U S A 2001. 98:6494-6499. [DOD] [CrossRef]
- Corella D, Qi L, Sorli JV, Godoy D, Portoles O, Coltell O, Greenberg AS, Ordovas JM. Obese subjects carrying the 11482GA polymorphism at the perilipin (PLIN) locus are resistant to weight loss following dietary energy restriction. J Clin Endocrinol Metab 2005. 90:5121-5126. [DOD] [CrossRef]
- Corella D, Qi L, Tai ES, Deurenberg-Yap M, Tan CE, Chew SK, Ordovas JM. Perilipin gene variation determines higher susceptibility to insulin resistance in Asian women when consuming a high-saturated fat, low-carbohydrate diet. Diabetes Care 2006. 29:1313-1319. [DOD] [CrossRef]
- Perez-Martinez P, Ordovas JM, Lopez-Miranda J, Gomez P, Marin C, Moreno J, Fuentes F, Fernandez de la Puebla RA, Perez-Jimenez F. Polymorphism exon 1 variant at the locus of the scavenger receptor class B type I gene: influence on plasma LDL cholesterol in healthy subjects during the consumption of diets with different fat contents. Am J Clin Nutr 2003. 77:809-813. [DOD]
- Osgood D, Corella D, Demissie S, Cupples LA, Wilson PW, Meigs JB, Schaefer EJ, Coltell O, Ordovas JM. Genetic variation at the scavenger receptor class B type I gene locus determines plasma lipoprotein concentrations and particle size and interacts with type 2 diabetes: the Framingham study. J Clin Endocrinol Metab 2003. 88:2869-2879. [DOD] [CrossRef]
- Perez-Martinez P, Perez-Jimenez F, Bellido C, Ordovas JM, Moreno JA, Marin C, Gomez P, Delgado-Lista J, Fuentes F, Lopez-Miranda J. A polymorphism exon 1 variant at the locus of the scavenger receptor class B type I (SCARB1) gene is associated with differences in insulin sensitivity in healthy people during the consumption of an olive oil-rich diet. J Clin Endocrinol Metab 2005. 90:2297-2300. [DOD] [CrossRef]
- Sweetser DA, Birkenmeier EH, Klisak IJ, Zollman S, Sparkes RS, Mohandas T, Lusis AJ, Gordon JI. The human and rodent fatty acid binding protein genes. A comparative analysis of their structure, expression and linkage relationships. J Biol Chem 1987. 262:16060-16071. [DOD]
- Bernlohr DA, Simpson MA, Hertzel AV, Banaszak L. Intracellular lipid-binding proteins and their genes. Annu Rev Nutr 1997. 17:277-303. [DOD] [CrossRef]
- Besnard P. Cellular and molecular aspects of fat metabolism in the small intestine. Proc Nutr Soc 1996. 5:19-37. [DOD]
- Van Nieuwenhoven FA, Var der Vusse GJ, Glatz JF. Membrane-associated and cytoplasmic fatty acid-binding proteins. Lipids 1996. 30:S223-S227. [DOD]
- Bass NM, Manning JA, Ockner RK, Gordon JI, Seetharam S, Alpers DH. Regulation of the biosynthesis of two distinct fatty acid-binding proteins in rat liver and intestine. J Biol Chem 1985. 260:1432-1436. [DOD]
- Ockner RK, Manning JA. Fatty acid binding protein in small intestine: identification, isolation and evidence for its role in cellular fatty acid binding transport. J Clin Invest 1974. 54:326-338. [DOD]
- Marin C, Perez-Jimenez F, Gomez P, Delgado J, Paniagua JA, Lozano A, Cortes B, Jimenez-Gomez Y, Gomez MJ, Lopez-Miranda J. The Ala54Thr polymorphism of the fatty acid-binding protein 2 gene is associated with a change in insulin sensitivity after a change in the type of dietary fat. Am J Clin Nutr 2005. 82:196-200. [DOD]
- Baier LJ, Sacchettini JC, Knowler WC, Eads J, Paolisso G, Tataranni PA, Mochizuki H, Bennett PH, Bogardus C, Prochazka M. An amino acid substitution in the human intestinal fatty acid binding protein is associated with increased fatty acid binding, increased fat oxidation, and insulin resistance. J Clin Invest 1995. 95:1281-1287. [DOD]
- Simopoulos AP. Evolutionary aspects of omega-3 fatty acids in the food supply. Prostaglandins Leukot Essent Fatty Acids 1999. 60(5-6):421-429. [DOD] [CrossRef]
- Panagiotakos DB, Pitsavos C, Polychronopoulos E, Chrysohoou C, Zampelas A, Trichopoulou A. Can a Meditteranean diet moderate the development and clinical progression of coronary heart disease? A systematic review. Med Sci Monit 2004. 10:RA193-RA198. [DOD]
- Tsimikas S, Philis-Tsimikas A, Alexopoulos S, Sigari F, Lee C, Reaven PD. LDL isolated from Greek subjects on a typical diet or from American subjects on an oleate-supplemented diet induce less monocyte chemotaxis and adhesion when exposed to oxidative stress. Arterioscler Thromb Vasc Biol 1999. 19:122-130. [DOD]
- Mata P, Alonso R, Lopez-Farre A, Ordovas JM, Lahoz C, Garces C, Caramelo C, Codoceo R, Blazquez E, de Oya M. Effect of dietary fat saturation on LDL oxidation and monocyte adhesion to human endothelial cells in vitro. Arterioscler Thromb Vasc Biol 1996. 16:1347-1355. [DOD]
- Sweetser DA, Birkenmeier EH, Klisak IJ, Zollman S, Sparkes RS, Mohandas T, Lusis AJ, Gordon JI. Oleic acid inhibits endothelial activation: a direct vascular antiatherogenic mechanism of a nutritional component of the Mediterranean diet. Arterioscler Thromb Vasc Biol 1999. 19:220-228. [DOD]
- Cutler JA, Follmann D, Allender PS. Randomized trials of sodium reduction: an overview. Am J Clin Nutr 1997. 65:643S-651S. [DOD]
- Sacks FM, Svetkey LP, Vollmer WM, Appel LJ, Bray GA, Harsha D, Obarzanek E, Conlin PR, Miller ER 3rd, Simons-Morton DG, et al. Effects on blood pressure of reduced dietary sodium and the dietary approaches to stop hypertension (DASH) diet. N Engl J Med 2002. 344:3-10. [DOD] [CrossRef]
- Tuck M, Corry D, Trujillo A. Salt-sensitive blood pressure and exaggerated vascular reactivity in the hypertension of diabetes mellitus. Am J Med 1991. 88:210-216. [DOD] [CrossRef]
This article has been cited by other articles:

|
Effects of Glimepiride on metabolic parameters and cardiovascular risk factors in patients with newly diagnosed type 2 diabetes mellitus
Xu DY, Zhao SP, Huang QX, Du W, Liu YH, Liu L, Xie XM
Diabetes Res Clin Pract 2010. 88(1):71-75
|
|

|
Evidence of interaction between type 2 diabetes susceptibility genes and dietary fat intake for adiposity and glucose homeostasis-related phenotypes
Ruchat SM, Elks CE, Loos RJ, Vohl MC, Weisnagel SJ, Rankinen T, Bouchard C, Perusse L
J Nutrigenet Nutrigenomics 2009. 2(4-5):225-234
|
|

|
Obesity and cardiovascular disease: From pathophysiology to risk stratification
Marinou K, Tousoulis D, Antonopoulos AS, Stefanadi E, Stefanadis C
Int J Cardiol 2009. Apr 25, epub ahead of print
|
|

|
Type 2 diabetes, multifactorial heredity and primary prevention
Bassan N, Vinuesa M, Soldano O, Sciarresi E, Bruera G, Di Sipio A, Venecia S
Acta Cient Estud 2008. 6(4):168-178
|
|

|
A genetic contribution to circulating cytokines and obesity in children
Cai G, Cole SA, Butte NF, Smith CW, Mehta NR, Voruganti VS, Proffitt JM, Comuzzie AG
Cytokine 2008. 44(2):242-247
|
|

|
Subclinical endothelial inflammation markers in a family with type I familial hyperaldosteronism caused by a de novo mutation
Stehr CB, Carvajal CA, Lacourt P, Alcaíno H, Mellado R, Cattani A, Mosso LM, Lavandera S, Fardella CE
Rev Med Chil 2008. 136(9):1134-1140
|
|

|
Age-dependent dichotomous effect of superoxide dismutase Ala16Val polymorphism on oxidized LDL levels
Dedoussis GV, Kanoni S, Panagiotakos DB, Louizou E, Grigoriou E, Chrysohoou C, Pitsavos C, Stefanadis C
Exp Mol Med 2008. 40(1):27-34
|
|

|
Impact of Metabolic Syndrome Risk Factors in First-Degree Relatives of Type 2 Diabetic Patients
Siewert S, Filipuzzi S, Codazzi L, Gonzalez I, Ojeda MS
Rev Diabet Stud 2007. 4(3):177-184
|
|

|
Genomics of Common Diseases
Revill P
Drug News Perspect 2007. 20(7):475-479
|
|
|


)
)
)

