Case Report
| Rev Diabet Stud,
2004,
1(4):193-197 |
DOI 10.1900/RDS.2004.1.193 |
A Case of Acquired Generalized Lipodystrophy with Cerebellar Degeneration and Type 2 Diabetes Mellitus
Pei-Jiuan Chao1, Jack C.-R. Tsai2, Dao-Ming Chang1, Shyi-Jang Shin3, Yau-Jiunn Lee2
1Department of Internal Medicine, Pingtung Christian Hospital, Pingtung, 90000 Taiwan.
2Department of Clinical Research, Pingtung Christian Hospital, Pingtung, 90000 Taiwan.
3Graduate Institute of Medicine, Kaohsiung Medical University, Kaohsiung, 80307 Taiwan.
Address correspondence to: Yau-Jiunn Lee, e-mail: t3275@ms25.hinet.net
Keywords: acquired generalized lipodydtrophy, diabetes mellitus, cerebellar degeneration
Abstract
Acquired generalized lipodystrophy (AGL) is a rare disorder of adipose tissue characterized by loss of fat from large regions of the body, occurring after birth. Its etiology remains unknown. Most AGL patients have had fasting and/or postprandial hyperinsulinemia, diabetes mellitus, hypertriglyceridemia, and fatty liver. We describe the case of a 30-year-old woman with a progressively unsteady gait and a generalized loss of body fat beginning at the age of 7. Cerebellar degeneration was revealed by imaging study, and the patient was eventually bedridden at the age of 15, due to progressive ataxia. She developed diabetes at the age of 25 without the presence of any evidence of ketoacidosis. The glutamic acid decarboxylase antibody was negative, C-peptide level 3.6 ng/ml, HbA1c 13%, triglyceride 412 mg/dl, total cholesterol 196 mg/dl, high-density lipoprotein-cholesterol 28 mg/dl, adiponectin 0.76 μg/ml, and resistin was 22.8 ng/ml at the initial state of diabetes. AGL accompanied by type 2 diabetes and cerebellar degeneration was diagnosed on the basis of the clinical features and metabolic derangements.
Introduction
It is well known that increasing adiposity is associated with insulin resistance and an increasing risk of type 2 diabetes mellitus (T2DM) [1]. There is evidence to support the role of deranged adipocyte metabolism and altered fat topography in the pathogenesis of glucose intolerance [2-4]. Failure to develop adequate adipose tissue mass, known as lipodystrophy, could also result in severe insulin resistance and diabetes. The insulin resistance in patients with lipodystrophy was thought to be the result of ectopic storage of lipid into the liver, skeletal muscle, and pancreatic β-cells [2]. Lipodystrophy in humans is an acquired or hereditary syndrome characterized by a decrease in adipose tissue mass [5]. The insufficient adipose tissue leads to excess energy storage as triglycerides in the liver and skeletal muscle and to insulin resistance and diabetes [2-4]. Recent advances in the understanding of the molecular basis of lipodystrophies have promoted understanding of how adipose tissue disorders can cause the metabolic syndrome and its complications [6, 7]. These hold promise for elucidating pathways and mechanisms by which common disorders of obesity cause metabolic complications.
The acquired form of lipodystrophy, initially reported by Ziegler in 1928 [8], is usually sporadic, although some familial cases have been reported [9]. Neurological problems in these patients are rare. We report on the case of a 30-year-old woman with acquired generalized lipodystrophy (AGL) who developed lipodystrophy and diabetes mellitus during childhood and adolescence. Furthermore, cerebellar ataxia with cerebellar degeneration developed in conjunction with lipodystrophy. Clinical course, therapeutic aspects, pathogenetic mechanisms, and possible links between adipopathy and insulin resistance are described and briefly reviewed.
Case report
The woman was born in 1975 and brought to our hospital in 2000 for the first time. The major complaints were polydipsia, polyuria and a weight loss of 2 kg in the past one and a half months. Disability in walking and standing and involuntary movements of the body were noted. A generalized loss of body fat over 4 extremities, the back, abdomen, face and neck, could be observed. No subcutaneous nodular swellings (panniculitis) had ever been found, but pressure sores over the scral areas had persisted for years. There was no acanthosis nigricans and hirsutism was absent. At the age of 30, the patient’s height was 138 cm; she weighed 30 kg.
Her mother stated that no abnormalities in growth or mental development had been noted until the girl was 7 years old. A series of examinations was performed, with an MRI scan inter alia disclosing cerebellar degeneration of unknown cause. No specific treatment had been given. Gait ataxia and involuntary movements of the body had deteriorated with time. At the age of 15, the patient eventually went through a bedridden period. Meanwhile, delayed and slow growth with insidious, progressive, extensive loss of subcutaneous fat over extremities, face, trunk and neck but without mental retardation were observed. The menarche was at the age of 15 and menstruation was irregular. Her family history proved negative for neurologic or hereditary disease, but one aunt had T2DM.
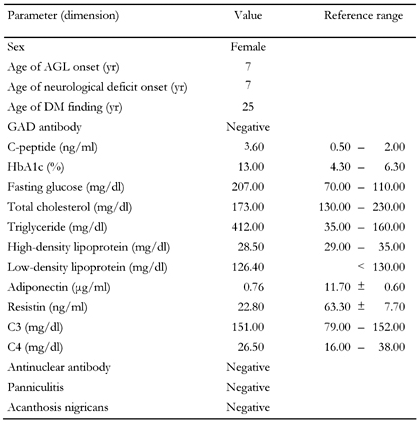
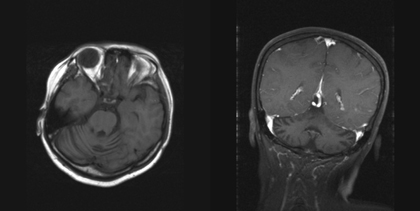
T2DM was diagnosed on the basis of elevated fasting plasma glucose, negative glutamic acid decarboxylase antibody, and no evidence of ketoacidosis. Her fasting C-peptide level was 3.6 ng/ml (0.5-2.0 ng/ml), HbA1c 13%, adiponectin 0.76 μg/ml (11.7 ± 0.6 μg/ml), and resistin 22.8 ng/ml (63.3 ± 7.7 ng/ml) at the initial state of diabetes. Blood biochemistry tests showed a triglyceride of 412 mg/dl, total cholesterol 196mg/dl, low-density lipoprotein 126 mg/dl, and high density lipoprotein 28 mg/dl. Antinulcear antibody was negative and C3 and C4 levels were within the reference ranges (Table 1). Her plasma thyroid hormones and thyroid-stimulating hormone levels were also within normal ranges. Abdominal sonography revealed fatty liver and atrophy of the uterus. A brain MRI scan showed relative prominence of bilateral cerebellar cortical sulci (Figure 1). Treatment with thiazolidnediones (TZD), insulin and fibric acid derivatives were given. The fasting sugar was 100 to 140 mg/dl, triglyceride 114 mg/dl, and HbA1c 8.5 to 9.1 % after treatment.
Table
1.
Clinical characteristics and metabolic parameters of the case reported |
|
 |
 |
Legend:
AGL: acquired generalized lipodystrophy, GAD: glutamic acid decarboxylase, HbA1c: glycated hemoglobin A1c, C3: serum level of CH50, complement 3, C4: serum level of CH50, complement 4. |
|
Discussion
The diagnostic criteria for AGL was proposed by Lawrence in 1946 [10] as 1. a generalized absence of fat, 2. insulin-resistant diabetes mellitus, 3. an absence of ketosis, 4. an elevated basal metabolic rate, and 5. severe hyperlipidemia with consequent hepatomegaly. Recently, Garg and Misra [9] subclassified AGL into three varieties: the panniculitis variety (type 1), the autoimmune diseases variety (type 2), and the idiopathic variety (type 3), which affect nearly 25%, 25%, and 50% of patients, respectively. The fat loss of AGL typically occurs during childhood and adolescence, affecting large areas of the body. Approximately one third of the patients have acanthosis nigricans, and hirsutism may be observed in women. Menstrual irregularity and polycystic ovarian syndrome may occur. Most AGL patients had fasting and/or postprandial hyperinsulinemia, diabetes mellitus, hypertriglyceridemia, and low serum levels of HDL-cholesterol, leptin, and adiponectin [9]. The patient in our case study shows a generalized lack of body fat with non-ketotic diabetes mellitus, hypertriglyceridemia, fatty liver, compatible with the clinical diagnosis of AGL. However, the present case may be an exceptional type of AGL, due to the accompanied neurological deficit as cerebellar degeneration. To date, AGL is a rare syndrome with less than 100 cases reported [9-18].
 |
|
Figure 1. MRI images of the brain. The axial and coronal section (T1WI) showed relative prominence of bilateral cerebellar cortical sulci. |
|
One case of familial lipodystrophy associated with neurodegeneration presented with spastic-ataxic gait and lower extremity paresthesiae at the age of 18 was observed before [19]. In general, no neurological abnormalities were found in previous cases of AGL. Chronic cerebellar degeneration may be related to alcoholism, drug abuse, hypothyroidism, paraneoplastic syndrome, heredity, Creutzfeldt-Jakob disease, posterior fossa tumors or malformations, celiac disease, or multiple sclerosis [20, 21]. Nonhereditary idiopathic cerebellar ataxia, or multiple system atrophy (MSA), is a sporadic adult-onset disease characterized by neurodegeneration in the basal ganglia, brain stem, cerebellum, and intermediolateral cell columns of the spinal cord. However, no apparent cause of cerebellar degeneration was found in the case reported here, and there were insufficient features compatible with the criteria for MSA [22].
The pathogenesis of AGL is not clear. Some AGL patients with or without panniculitis have clinical or serological evidence of autoimmune diseases [5]. Shill et al. observed a high prevalence of antiganglioside antibodies in patients with cerebellar degeneration [23]. The present case of AGL with cerebellar degeneration showed no clinical evidence of autoimmune diseases. If the association of autoimmunity contributes to fat loss and neurological dysfunction, and this could be demonstrated, it may lead to an effective therapy for these conditions.
Recent studies have shown recombinant leptin to be effective in improving hyperglycemia, dyslipidemia, and hepatic steatosis in patients with lipodystrophies [24-26]. However, leptin therapy remains tentatively. TZD, an insulin-sensitizing agent has proved to increase the plasma adiponectin level, increase adipocyte differentiation and proliferation, and decrease insulin resistance, implying potential use in the management of lipodystrophy [27, 28]. Treatment with insulin-sensitizing agents such as TZD has improved the glycated hemoglobuin A1c level. Whether TZD has advantages in the treatment of patients with lipodystrophy needs to be clarified in a large-scale study.
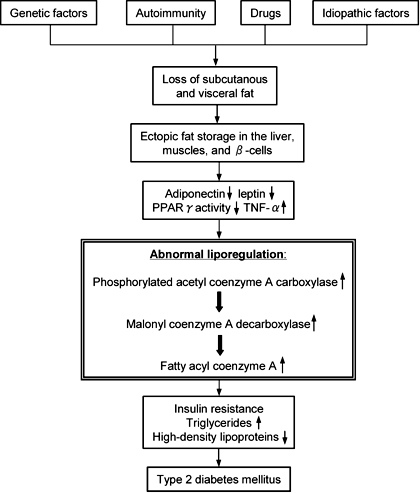
Lipodystrophy in humans is a syndrome characterized by a decrease in adipose tissue mass. The insufficient adipose tissue mass leads to chronically elevated plasma free fatty acid (FFA) and excess energy storage as triglyceride in the liver and skeletal muscle. Long-term elevated plasma FFA results in an impaired β-cell response to glucose [29], and insulin resistance in muscle and liver [2-4]. The inadequate adipose tissue mass leads to ectopic fat storage and predisposes to the development of insulin resistance. Serum leptin and adiponectin levels reflecting the amount of body fat are extremely low in patients with AGL and may be related to severe insulin resistance and its metabolic complications in lipodystrophies [11, 13, 30, 31]. The plasma resistin is low in those patients, as reported by Iglesias and his colleagues [18], indicating that plasma resistin may not be related to the insulin resistance in patients with lipodystrophy. Tumor necrosis factor-a, a proinflammatory cytokine, also secreted from adipocytes, is reported to elevate plasma levels in a patient with AGL [18] and it is assumed that this might play a role in insulin resistance. The possible pathological mechanisms linking lipodystrophy and insulin resistance are summarized in Figure 2.
 |
|
Figure 2. Possible pathogenetic mechanisms of type 2 diabetes mellitus in lipodystrophy. (PPARγ: peroxisomal proliferators-activated receptor γ, TNF-α: tumor necrosis factor-α). |
|
Conclusion
This report shows an uncommen case in which AGL is accompanied by degenerative cerebellar disorder. The insufficient adipose tissue mass leads to excess energy storage in ectopic fat storage organs that finally results in insulin resistance and diabetes.
References
- Smith DO, LeRoith D. Insulin resistance syndrome, pre-diabetes, and the prevention of type 2 diabetes mellitus. Clin Cornerstone 2004. 6:7-6. [DOD] [CrossRef]
- Ravussin E, Smith SR. Increased fat intake, impaired fat oxidation, and failure of fat cell proliferation result in ectopic fat storage, insulin resistance, and type 2 diabetes mellitus. Ann N Y Acad Sci 2002. 967:363-378. [DOD]
- Bays H, Mandarino L, DeFronzo RA. Role of the adipocyte, free fatty acids, and ectopic fat in pathogenesis of type 2 diabetes mellitus: peroxisomal proliferator-activated receptor agonists provide a rational therapeutic approach. J Clin Endocrinol Metab 2004. 89:463-478. [DOD] [CrossRef]
- Lewis GF, Carpentier A, Adeli K, Giacca A. Disordered fat storage and mobilization in the pathogenesis of insulin resistance and type 2 diabetes. Endocr Rev 2002. 23:201-229. [DOD] [CrossRef]
- Garg A. Lipodystrophies. Am J Med 2000. 108:143-152. [DOD]
- Garg A, Misra A. Lipodystrophies: rare disorders causing metabolic syndrome. Endrocrinol Metab Clin N Am 2004. 33:305-331. [DOD] [CrossRef]
- Garg A. Regional adiposity and insulin resistance. J Clin Endocrinol Metab 2004. 89:4206-4210. [DOD] [CrossRef]
- Ziegler LH. Lipodystrophies: report of seven cases. Brain 1928. 51:145-167. [DOD]
- Misra A, Garg A. Clinical features and metabolic derangements in acquired generalized lipodystrophy: case reports and review of the literature. Medicine 2003. 82:129-146. [DOD] [CrossRef]
- Lawrence RD. Lipodystrophy and hepatomegaly with diabetes, lipemia, and other metabolic disturbances. Lancet 1946. 1:724-731. [DOD] [CrossRef]
- Pardini VC, Victoria IM, Rocha SM, Andrade DG, Rocha AM, Pieroni FB, Milagres G, Purisch S, Velho G. Leptin levels, beta-cell function, and insulin sensitivity in families with congenital and acquired generalized lipoatropic diabetes. J Clin Endocrinol Metab 1998. 83:503-508. [DOD] [CrossRef]
- Garg A. Acquired and inherited lipodystrophies. N Engl J Med 2004. 350:1220-1234. [DOD] [CrossRef]
- Haque WA, Shimomura I, Matsuzawa Y, Garg A. Serum adiponectin and leptin levels in patients with lipodystrophies. J Clin Endocrinol Metab 2002. 87:2395-2398. [DOD] [CrossRef]
- Sarai T, Kawanishi K, Saito Y, Aoi K, Nishina Y, Ofuji T. Lipoatrophic diabetes. Report of a case. Act Med Okayama 1978. 32:309-318. [DOD]
- Vantyghem MC, Vigouroux C, Magre J, Desbois-Mouthon C, Pattou F, Fontaine P, Lefebvre J, Capeau J. Late-onset lipoatrophic diabetes. Phenotypic and genotypic familial studies and effect of treatment with metformin and lispro insulin analog. Diabetes Care 1999. 22:1374-1376. [DOD]
- Panz VR, Wing JR, Raal FJ, Kedda MA, Joffe BI. Improved glucose tolerance after effective lipid-lowering therapy with bezafibrate in a patient with lipoatrophic diabetes mellitus: a putative role for Randle's cycle in its pathogenesis? Clin Endocrinol 1997. 46:365-368. [DOD]
- Hubler A, Abendroth K, Keiner T, Stocker W, Kauf E, Hein G, Stein G. Dysregulation of insulin-like growth factors in a case of generalized acquired lipoatrophic diabetes mellitus (Lawrence Syndrome) connected with autoantibodies against adipocyte membranes. Exp Clin Endocrinol Diabetes 1998. 106:79-84. [DOD]
- Iglesias P, Fidalgo PA, Codoceo R, Diez JJ. Lipoatrophic diabetes in an elderly woman: Clinical course and serum adipocytokine concentrations. Endrocrine Journal 2004. 51:279-286. [DOD] [CrossRef]
- Berger JR, Oral EA, Taylor SI. Familial lipodystrophy associated with neurodegeneration and congenital cataracts. Neurology 2002. 58:43-47. [DOD] [CrossRef]
- Aminoff MJ, Greeberg DA, Simon RP. Clinical Neurology. Prentice-Hall International 1996. Chapter 4, pp. 110-120. [DOD]
- Cooke WT, Thomas-Smith W. Neurological disorders associated with adult celiac disease. Brain 1966. 89:683-722. [DOD]
- Wenning GK, Colosimo C, Feser F, Poewe W. Multiple system atrophy. Lancet Neurol 2004. 3:93-103. [DOD] [CrossRef]
- Shill HA, Alaedini A, Latov N, Hallett M. Anti-ganglioside antibodies in idiopathic and hereditary cerebellar degeneration. Neurology 2003. 60:1672-1673. [DOD]
- Oral EA, Simha V, Ruiz E, Andewelt A, Premkumar A, Snell P, Wagner AJ, DePaoli AM, Reitman ML, Taylor SI, et al. Leptin-replacement therapy for lipodystrophy. N Engl J Med 2002. 346:570-578. [DOD] [CrossRef]
- Van Gaal LF, Mertens IL, Abrams PJ. Health risks of lipodystrophy and abdominal fat accumulation: therapeutic possibilities with leptin and human growth hormone. Growth Horm IGF Res 2003. 13(Suppl A):S4-S9. [DOD] [CrossRef]
- Petersen KF, Oral EA, Dufour S, Befroy D, Ariyan C, Yu C, Cline GW, DePaoli AM, Taylor SI, Gorden P, et al. Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J Clin Invest. 2002. 109:1345-1350. [DOD]
- Savage DB, Tan GD, Acerini CL, Jebb SA, Agostini M, Gurnell M, Williams RL, Umpleby AM, Thomas EL, Bell JD, et al. Human metabolic syndrome resulting from dominant-negative mutations in the nuclear receptor peroxisome proliferator-activated receptor-gamma. Diabetes 2003. 52:910-917. [DOD]
- Arioglu E, Duncan-Morin J, Sebring N, Rother KI, Gottlieb N, Lieberman J, Herion D, Kleiner DE, Reynolds J, Premkumar A, et al. Efficacy and safety of troglitazone in the treatment of lipodystrophy syndromes. Ann Intern Med 2000. 133:263-274. [DOD]
- McGarry JD. Banting lecture 2001: dysregulation of fatty acid metabolism in the etiology of type 2 diabetes. Diabetes 2002. 51:7-18. [DOD]
- Reitman ML, Arioglu E, Gavrilova O, Taylor SI. Lipoatrophy revisited. Trends Endocrinol Metab 2000. 11:410-416. [DOD] [CrossRef]
- Weyer C, Funahashi T, Tanaka S, Hotta K, Matsuzawa Y, Pratley RE, Tataranni PA. Hypoadiponectinemia in obesity and type 2 diabetes: close association with insulin resistance and hyperinsulinemia. J Clin Endocrinol Metab 2001. 86:1930-1935. [DOD]
This article has been cited by other articles:
|


)
)
)


