Original Data
| Rev Diabet Stud,
2009,
6(2):97-103 |
DOI 10.1900/RDS.2009.6.97 |
Type 1 Diabetes Development Requires Both CD4+ and CD8+ T cells and Can Be Reversed by Non-Depleting Antibodies Targeting Both T Cell Populations
Jenny M. Phillips, Nicole M. Parish, Tim Raine, Chris Bland, Yvonne Sawyer, Hugo De La Peña, Anne Cooke
Department of Pathology, University of Cambridge, Tennis Court Rd., Cambridge, CB21 QP, United Kingdom
Address correspondence to: Anne Cooke, e-mail: ac@mole.bio.cam.ac.uk
Manuscript submitted July 15, 2009; resubmitted July 27, 2009; accepted August 4, 2009.
Keywords: type 1 diabetes, T cell, CD4+, CD8+, NOD, NOD.scid, pancreas infiltration, CD8 alpha chain, dendritic cell, IgG2 antibody, aCD3 antibody
Abstract
Type 1 diabetes development in NOD mice appears to require both CD4+ and CD8+ T cells. However, there are some situations where it has been suggested that either CD4+ or CD8+ T cells are able to mediate diabetes in the absence of the other population. In the case of transgenic mice, this may reflect the numbers of antigen-specific T cells able to access the pancreas and recruit other cell types such as macrophages leading to a release of high concentrations of damaging cytokines. Previous studies examining the requirement for CD8+ T cells have used antibodies specific for CD8α. It is known that CD8α is expressed not only on αβ T cells, but also on other cell types, including a DC population that may be critical for presenting islet antigen in the pancreatic draining lymph nodes. Therefore, we have re-examined the need for both CD4+ and CD8+ T cell populations in diabetes development in NOD mice using an antibody to CD8β. Our studies indicate that by using highly purified populations of T cells and antibodies specific for CD8+ T cells, there is indeed a need for both cell types. In accordance with some other reports, we found that CD4+ T cells appeared to be able to access the pancreas more readily than CD8+ T cells. Despite the ability of CD4+ T cells to recruit CD11b class II positive cells, diabetes did not develop in the absence of CD8+ T cells. These studies support the observation that CD8+ T cells may be final effector cells. As both T cell populations are clearly implicated in diabetes development, we have used a combination of non-depleting antibodies to target both CD4-positive and CD8-positive cells and found that this antibody combination was able to reverse diabetes onset in NOD mice as effectively as anti-CD3 antibodies.
Introduction
There is much evidence to suggest that CD8+ T cells play a role in the development of diabetes. Early studies in NOD mice showed that the transfer of diabetes by spleen cells from diabetic donors into immuno-compromised recipients required the presence of both CD4+ and CD8+ T cells [1-3]. Further studies in NOD mice showed that CD8+ T cells were required for cyclophosphamide-induced diabetes [4] and also that MHC class I expression was required in NOD mice for diabetes to spontaneously develop [5, 6]. Depletion of CD8+ T cells has been shown to afford protection from disease and an overall reduction in islet infiltration [2, 7], which has led to the proposition that CD8+ T cells may facilitate recruitment of lymphocytes to the pancreas. This interpretation is supported by the findings of Wang et al. who showed that early administration of an IgG2a rat anti-mouse CD8 monoclonal antibody prevents insulitis in NOD mice, with no effect of such treatment at later time points [8]. However, the requirement for CD8+ T cells in the development of diabetes in NOD mice was not a universal observation. It was suggested that CD4+ T cells alone could transfer diabetes to NOD.scid recipients, if they were derived from a diabetic donor, but that CD8+ T cells were required if the CD4 population was derived from pre-diabetic mice [9].
Every in vivo antibody study targeting CD8+ T cells has used antibodies directed at CD8α. The antibodies used to deplete CD8+ T cells in vitro have also been directed against the CD8α chain. There are several cell types apart from αβ T cells that express CD8α including γδ T cells, NKT cells, and some dendritic cells (DCs). In these cases the αα homodimer is expressed. This means that all previous studies could not distinguish between effects on αβ T cells and on other cell types. As CD8α-expressing DCs have been shown to play a role in cross presentation, a process of particular importance in the presentation of islet antigens and T cell activation in the pancreatic draining lymph node [10], we felt it important to clearly establish that αβ T cell depletion alone influenced diabetes onset. We have used an antibody to the CD8β chain to show that depletion of CD8+ cells with this antibody prevents diabetes development in a transfer model of T1D in the NOD mouse. This confirmed that CD8+ T cells are indeed required for diabetes development in NOD mice.
We have previously shown that administration of a short course of non-depleting anti-CD4 antibody to 6 week old NOD mice provides long term prevention from diabetes development [11]. However, this antibody was unable to reverse diabetes onset once it was established; unlike anti-CD3 which had been shown to reverse diabetes onset in NOD mice [12]. As the anti-CD3 antibody would be able to target both CD4+ T cells and CD8+ T cells, we carried out a series of experiments to establish whether the use of anti-CD4 antibodies together with anti-CD8 antibodies could reverse diabetes onset. For these studies we used both a non-depleting anti-CD4 as well as a non-depleting anti-CD8 antibody. The latter antibody recognized CD8α. For therapeutic purposes, using in vivo antibody treatment with anti-CD8 antibodies, there may be a significant advantage in using an antibody that also targets other cell types such as CD8α DCs. Such an antibody may be able to target not only the T cells, but also the cells involved in cross-presenting islet antigens in the pancreatic draining lymph nodes.
Materials and methods
Mice
NOD mice were housed and bred under specific pathogen-free conditions in the Pathology Department, University of Cambridge animal facilities. NOD.scid mice were maintained in microisolator cages with filtered air and handled under sterile conditions in a laminar flow hood. All animal work was carried out under UK Home Office project licence regulations after approval by the Ethical Review Committee of the University of Cambridge.
Antibodies and in vivo treatment
The following hybridomas were a gift from Herman Waldmann (Oxford, UK): YTS 177.9.6.1 (rat IgG2a, anti-CD4), YTS 105.18.10 (rat IgG2a, anti-CD8α), YTS 191.1.2 (rat IgG2b, anti-CD4), YTS 169.4.2 (rat IgG2b, anti-CD8α), YTS 156.7.7 (rat IgG2b, anti-CD8β), and the isotype control hybridoma YFC 51 (rat IgG2b). The isotype control hybridoma MAC 219 (rat IgG2a) was a gift from Geoff Butcher (Babraham, UK). All hybridomas were grown in our own laboratory in hollow fibre cartridges. Antibodies were purified by precipitation with 50% saturated ammonium sulphate and dialyzed extensively against PBS. An estimate of total protein was determined from the OD280. Antibody concentrations were determined by an anti-rat immunoglobulin ELISA. The endotoxin levels were <1EU/mg protein and the preparations were stored at -20°C until use. Groups of mice were treated intra-peritoneally (i.p.) with the test antibodies or isotype controls. Timings of antibody administrations are given in the results section or figure legends. Mice were tested for the presence of urinary glucose using Diastix (Bayer plc, Newbury, UK) test strips. Mice showing positive urinary glucose on at least two occasions were considered diabetic and this was confirmed using a Glucometer Ascensia Esprit 2 (Bayer plc, Newbury, UK).
The incidence of diabetes in females in our NOD colony is currently 80-90% by 30 weeks of age. Diabetic female mice which were treated with antibodies were regularly tested for blood glucose in whole blood from the tail vein.
T cell isolation
CD4 and CD8 T cells were isolated using anti-CD4 or anti-CD8 magnetic beads (Miltenyi Biotec, Surrey UK) used according to the manufacturer's instructions. NOD donors of the T cells were pretreated with 3 intraperitoneal injections on days -7, -5 and -3 with either a depleting anti-CD4 (YTS 191), for the CD8 cells or a depleting anti-CD8 (YTS 169), for the CD4 cells. This method gave populations of CD4 or CD8 cells, which had less than 0.5% contamination with the other cell population (data not shown). T cells were injected intravenously into NOD.scid recipients.
Immunohistochemistry
Pancreases were removed after sacrifice and snap frozen in isopentane. Five-micrometer cryostat sections were air-dried and fixed in acetone for 10 minutes. Air-dried sections were stored at -80°C. Pancreatic β-cells were detected by pre-blocking sections with 20% NMS followed by incubation with guinea pig anti-porcine insulin (Dako, High Wycombe, GB) in 10% NMS and detected by rhodaminated goat anti-guinea-pig IgG (ICN Pharmaceuticals, Thame, GB) in 10% NMS. CD4 was detected using monoclonal antibody supernatant KT4 from Prof. K. Tomonari, (Japan), CD8β with antibody supernatant YTS 156, and CD11b with M 1/70 (BD Pharmingen) in 10% NMS and visualized with FITC goat anti-rat Ig (Serotec, Kidlington, GB). Sections were photographed using a Zeiss Axiophot photomicroscope at magnification ×40.
Results
Diabetes transfer into immuno-compromised recipients was inhibited by anti-CD8β
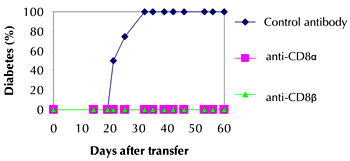
When spleen cells from diabetic NOD donors were transferred into immuno-compromised recipient mice, diabetes developed usually within 2-6 weeks. From Figure 1 it can be seen that when NOD.scid recipients of 2×107 spleen cells from a pool of diabetic donors were treated with either anti-CD8α or anti-CD8β, diabetes development was completely prevented. Control antibody treatment had no effect with 100% of recipient mice developing diabetes within 3 weeks of cell transfer. This shows that it is indeed the CD8+ T cells that are required together with CD4+ T cells for diabetes transfer.
 |
 |
Figure 1. Diabetes transfer is inhibited by anti-CD8α and anti-CD8β. Spleen cells from a pool of diabetic NOD donors (2x107) were transferred intravenously to NOD.scid recipients. The recipient mice (4 per group) were treated with 2 mg of the appropriate antibody i.p. at the same time as the cell transfer, and also on days 2 and 5 after the transfer. Mice were tested every 2-3 days for the presence of urinary glucose and considered diabetic when positive on at least two occasions. |
|
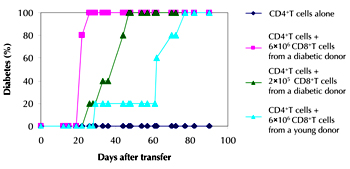
Transfer of 1x107 highly purified CD4+ T cells from diabetic donor mice failed to transfer diabetes, while addition of 6x106 purified CD8+ T cells from a diabetic donor to this CD4+ T cell population resulted in 100% transfer of disease within 3 weeks. When fewer purified CD8+ T cells (2x105) from a diabetic donor were co-transferred with the highly pure CD4+ T cells, disease transfer was slightly delayed. This suggests that if a CD4+ T cell preparation was transferred with a small contaminant of CD8+ T cells these could expand up over time and mediate diabetes transfer.
If 6x106 purified CD8+ T cells were derived from a 4 week old non-diabetic NOD mouse, their addition to the CD4+ T cell population resulted in a much delayed diabetes transfer (Figure 2). These studies suggest that the frequency of islet-reactive CD8+ T cells is reduced in non-diabetic younger mice and that CD8+ T cells are primed to islet antigen and expand during diabetes development in NOD mice.
 |
|
Figure 2. Purified CD4 T cells only transfer diabetes to NOD.scid recipients when CD8 cells are present. Purified CD4 cells (1x107) were injected i.v. into NOD.scid recipients alone (n = 5), together with a high number of purified CD8 cells from diabetic donors (6x107, n = 5), together with a low number of purified CD8 cells from diabetic donors (2x107, n = 5), or together with purified CD8 cells from 4 week old female donors (6x107, n = 5). Mice were tested every 2-3 days for the presence of urinary glucose and considered diabetic when positive on at least two occasions. |
|
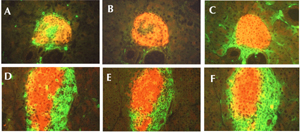
Immunohistochemical analysis of the recipient pancreas 14 days after transfer of the highly purified T cell populations showed that following transfer of CD4+ T cells, the transferred cells could be found at islet sites in the pancreas. There was also recruitment of CD11b-positive cells (macrophages and dendritic cells) with class-II-positive and F4/80-positive cells (Figure 3 A, C and data not shown). As expected with this transfer, there were no CD8β+ T cells (Figure 3B).
In contrast to the results obtained with the purified CD4+ T cell transfers, none of the transferred T cells could be seen in the pancreas following purified CD8+ T cell transfers. This is an interesting result given that the transferred T cells were clearly present in the pancreatic lymph nodes and there was no recruitment of class II positive cells around the islet area (data not shown). When both purified populations were transferred together, CD4, CD8 and CD11b cells, were all seen at the islet sites (Figure 3D-F).
 |
|
Figure 3. Purified CD4 T cells transferred to NOD.scid recipients do not cause diabetes but they do enter the pancreas and recruit CD11b cells. Purified CD4 cells (1x107) or purified CD8 cells (6x107) were injected i.v. into NOD.scid recipients either individually or combined. After 14 days, sequential sections of pancreas were examined (top row received CD4 cells alone and bottom row received CD4 and CD8 cells). A and D: Sections were stained for insulin (red) together with CD4 (green). B and E: Sections stained for CD8β (green). C and F: Sections stained for CD11b (green). |
|
Combined treatment with anti-CD4 and anti-CD8 can reverse diabetes in NOD mice
As both CD4+ T cells and CD8+ T cell populations are involved in the development of diabetes, we examined whether targeting both of these can reverse established diabetes at onset. We have previously shown that a non-depleting anti-CD4 antibody was able to prevent the spontaneous development of diabetes in NOD mice, if administered for two weeks when mice were 6 weeks of age; but was unable to reverse ongoing diabetes [11]. One interpretation of this result is that at this stage of the disease it becomes necessary to additionally target the CD8 population. An advantage of the non-depleting rat monoclonal anti-CD4 antibody is that the antibody induces tolerance to its own rat Fc region and does not generate an anti-globulin response. When this antibody was used in combination with the IgG2a anti-CD8α antibody, YTS105, CD8+ T cells were not depleted, but were functionally inactivated. These antibodies have previously been used in combination to induce alloantigen-specific tolerance of skin, cardiac and bone marrow allografts [13-16].
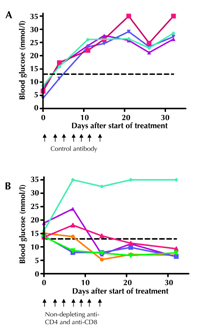
Female NOD mice were tested for diabetes onset and once tested positively they were administered a combination of two non-depleting antibodies, an anti-CD4 and an anti-CD8α. In Figure 4, it can be seen that the combination therapy was able to reverse diabetes onset. In contrast, diabetes progressed inexorably in mice given isotype control antibody. The reversal of diabetes observed with combined anti-CD4 and anti-CD8 is comparable to that obtained following treatment with anti-CD3 [12, 17].
 |
|
Figure 4. Remission of diabetes by treatment of NOD mice with non-depleting anti-CD4 and anti-CD8. At the time of diagnosis of diabetes by high urinary glucose, NOD mice were treated with either the IgG2a isotype control (MAC 219) (A), or a combination of non-depleting antibodies to CD4 (YTS 177) and CD8 (YTS 105) (B). Antibodies (2 mg of MAC 219, or 1 mg each of YTS 177 and YTS 105) were given i.p. every other day for 2 weeks. The glucose levels in the blood of individual mice treated with the antibodies are shown. Mice with glucose values above the dotted black line (13 mmol/l glucose) are considered diabetic. |
|
Discussion
There are many studies suggesting that diabetes development in NOD mice requires the presence of both CD4+ and CD8+ T cells. Some of these data suggest that there is a requirement for both populations to be present to transfer diabetes into immuno-compromised recipients [2, 7]. On the other hand, there are also data suggesting that CD4+ T cells from diabetic NOD donors may be able to transfer disease on their own [9].
We and others using monoclonal anti-CD8 antibodies have used antibodies that target CD8α. However, as CD8α is also expressed on DCs and γδ T cells, we have carried out a series of experiments using a monoclonal antibody to CD8β to fully clarify the role of CD8+ T cells in diabetes development in NOD mice. Our studies indicated firstly that CD8+ T cells are required together with CD4+ T cells, for diabetes development. Secondly, our studies showed that in the absence of CD8+ T cells, CD4+ T cells infiltrate the pancreas and recruit CD11b+ cells to the islet area, but that diabetes nevertheless does not develop. In the absence of CD4+ T cells, there is no significant influx of cells into the islet area.
The data presented here is strongly supportive of the view that CD8+ T cells play an effector cell role [18, 19]. Our previous studies have shown that CD11b-positive cells are also required for the transfer of diabetes by spleen cells from diabetic donors [20], indicating a dependency on all three types of cells for optimal diabetes induction in NOD mice. There are several studies showing that diabetes can be transferred by either CD4+ or CD8+ T clones alone or by transgenic T cells generated using the clone TCRs [21-24]. However, in these cases a large bolus of islet-specific T cells were transferred. Once activated, these were capable of recruiting large numbers of CD11b-positive cells and mediating substantial aggressive inflammation in the pancreas. The development of diabetes in the NOD mouse is more measured. CD4+ T cells may play another role in facilitating the survival and activity of islet antigen-specific CD8+ T cells, possibly following activation in the pancreatic draining lymph node. Such an effect has been nicely demonstrated in a model system using transgenic mice expressing ovalbumin in the pancreas together with TCR transgenic T cells specific for ovalbumin [25].
Monoclonal antibodies have been employed with considerable success to treat a range of pathological conditions including autoimmunity. Such therapeutic antibodies have included those directed against T cell surface antigens as well as antibodies against co-stimulatory molecules and cytokines. The observation that anti-CD3 antibodies were able to reverse diabetes onset in NOD mice [12] was the basis upon which antibodies targeting human anti-CD3 were used to treat newly diagnosed patients [26, 27]. It had been hoped that, following tolerance induction, β-cell repair and/or regeneration might be able to ensue and replace the destroyed and impaired β-cell mass [28]. However, this has thus far not been realized, as anti-CD3 antibodies alone do not seem to facilitate this process, possibly because this agent inhibits the elaboration of molecules important to drive regeneration [17, 29]. Recent studies have shown improved β-cell recovery in diabetic NOD mice when they are given anti-CD3 combined with exendin-4 [30].
There are also data suggesting that anti-CD3, and the combined anti-CD4 and anti-CD8, might function in different ways. Despite Fc modification, the anti-CD3 antibody causes some transient side effects in patients, which may relate to cytokine release [27]. Comparable studies using an aglycosyl anti-CD3 in the NOD mouse have not been reported, but it might be anticipated that this would also cause some cytokine release, albeit less than the conventional anti-CD3 antibody. Whether such cytokine release is also seen with anti-CD4 and anti-CD8 antibody treatments remains to be clarified. Both anti-CD3 and non-depleting anti-CD4 have been shown to induce regulatory T cells, which may play a key role in maintaining tolerance.
Tolerance induced by non-depleting anti-CD8 antibodies is dependent on IL-10 [11], while that induced by anti-CD3 and anti-CD4 has been reported to be TGFβ-dependent [31, 32]. A more detailed understanding of the mechanism by which these therapeutic approaches mediate diabetes reversal, should lead to an identification of key pathways that could be targeted. This could lead to the development of small molecule inhibitors that could favor both inhibition of the autoreactive process and β-cell recovery, and might circumvent the development of the side effects associated thus far with some of the antibody treatments.
Conflict of interest statement: The authors declare that they have no conflict of interests.
Acknowledgments:
This research was funded by Diabetes UK and The Wellcome Trust.
References
- Hutchings PR, Cooke A. The transfer of autoimmune diabetes in NOD mice can be inhibited or accelerated by distinct cell populations present in normal splenocytes taken from young males. J Autoimmun 1990. 3:175-185. [DOD] [CrossRef]
- Miller BJ, Appel MC, O'Neil JJ, Wicker LS. Both the Lyt-2+ and L3T4+ T cell subsets are required for the transfer of diabetes in nonobese diabetic mice. J Immunol 1988. 140:52-58. [DOD]
- Yagi H, Matsumoto M, Kunimoto K, Kawaguchi J, Makino S, Harada M. Analysis of the roles of CD4+ and CD8+ T cells in autoimmune diabetes of NOD mice using transfer to NOD athymic nude mice. Eur J Immunol 1992. 22(9):2387-2393. [DOD] [CrossRef]
- Charlton B, Bacelj A, Mandel TE. Administration of silica particles or anti-Lyt2 antibody prevents beta-cell destruction in NOD mice given cyclophosphamide. Diabetes 1988. 37:930-935. [DOD] [CrossRef]
- Hamilton-Williams EE, Serreze DV, Charlton B, Johnson EA, Marron MP, Mullbacher A, Slattery RM. Transgenic rescue implicates beta2-microglobulin as a diabetes susceptibility gene in nonobese diabetic (NOD) mice. Proc Natl Acad Sci U S A 2001. 98(20):11533-11538. [DOD] [CrossRef]
- Serreze DV, Gallichan WS, Snider DP, Croitoru K, Rosenthal KL, Leiter EH, Christianson GJ, Dudley ME, Roopenian DC. MHC class I-mediated antigen presentation and induction of CD8+ cytotoxic T-cell responses in autoimmune diabetes-prone NOD mice. Diabetes 1996. 45(7):902-908. [DOD] [CrossRef]
- Hutchings PR, Simpson E, O'Reilly LA, Lund T, Waldmann H, Cooke A. The involvement of Ly2+ T cells in beta cell destruction. J Autoimmun 1990. 3(Suppl 1):101-109. [DOD]
- Wang B, Gonzalez A, Benoist C, Mathis D. The role of CD8+ T cells in the initiation of insulin-dependent diabetes mellitus. Eur J Immunol 1996. 26:1762-1769. [DOD] [CrossRef]
- Christianson SW, Shultz LD, Leiter EH. Adoptive transfer of diabetes into immunodeficient NOD-scid/scid mice. Relative contributions of CD4+ and CD8+ T-cells from diabetic versus prediabetic NOD.NON-Thy-1a donors. Diabetes 1993. 42:44-55. [DOD] [CrossRef]
- Kurts C, Miller JF, Subramaniam RM, Carbone FR, Heath WR. Major histocompatibility complex class I-restricted cross-presentation is biased towards high dose antigens and those released during cellular destruction. J Exp Med 1998. 188(2):409-414. [DOD] [CrossRef]
- Parish NM, Bowie L, Zusman Harach S, Phillips JM, Cooke A. Thymus-dependent monoclonal antibody-induced protection from transferred diabetes. Eur J Immunol 1998. 28(12):4362-4373. [DOD] [CrossRef]
- Chatenoud L, Thervet E, Primo J, Bach JF. Anti-CD3 antibody induces long-term remission of overt autoimmunity in nonobese diabetic mice. Proc Natl Acad Sci U S A 1994. 91:123-127. [DOD] [CrossRef]
- Chen Z, Cobbold S, Metcalfe S, Waldmann H. Tolerance in the mouse to major histocompatibility complex-mismatched heart allografts, and to rat heart xenografts, using monoclonal antibodies to CD4 and CD8. Eur J Immunol 1992. 22:805-810. [DOD] [CrossRef]
- Cobbold SP, Martin G, Waldmann H. The induction of skin graft tolerance in major histocompatibility complex-mismatched or primed recipients: primed T cells can be tolerized in the periphery with anti-CD4 and anti-CD8 antibodies. Eur J Immunol 1990. 20:2747-2755. [DOD] [CrossRef]
- Qin S, Cobbold SP, Pope H, Elliott J, Kioussis D, Davies J, Waldmann H. "Infectious" transplantation tolerance. Science 1993. 259(5097):974-977. [DOD] [CrossRef]
- Qin SX, Wise M, Cobbold SP, Leong L, Kong YC, Parnes JR, Waldmann H. Induction of tolerance in peripheral T cells with monoclonal antibodies. Eur J Immunol 1990. 20(12):2737-2745. [DOD] [CrossRef]
- Phillips JM, O'Reilly L, Bland C, Foulis AK, Cooke A. Patients with chronic pancreatitis have islet progenitor cells in their ducts, but reversal of overt diabetes in NOD mice by anti-CD3 shows no evidence for islet regeneration. Diabetes 2007. 56(3):634-640. [DOD] [CrossRef]
- Kay TW, Dudek NL, Graham K, Estella E, Angstetra E, McKenzie MD, Allison J, Thomas HE. Cytotoxic T cell mechanisms of beta cell destruction in non-obese diabetic mice. Novartis Found Symp 2008. 292:68-78. [DOD] [CrossRef]
- Thomas HE, Kay TW. Beta cell destruction in the development of autoimmune diabetes in the non-obese diabetic (NOD) mouse. Diabetes Metab Res Rev 2000. 16:251-261. [DOD] [CrossRef]
- Hutchings P, Rosen H, O'Reilly L, Simpson E, Gordon S, Cooke A. Transfer of diabetes in mice prevented by blockade of adhesion-promoting receptor on macrophages. Nature 1990. 348(6302):639-642. [DOD] [CrossRef]
- Bradley BJ, Haskins K, La Rosa FG, Lafferty KJ. CD8 T cells are not required for islet destruction induced by a CD4+ islet-specific T-cell clone. Diabetes 1992. 41:1603-1608. [DOD] [CrossRef]
- Graser RT, DiLorenzo TP, Wang F, Christianson GJ, Chapman HD, Roopenian DC, Nathenson SG, Serreze DV. Identification of a CD8 T cell that can independently mediate autoimmune diabetes development in the complete absence of CD4 T cell helper functions. J Immunol 2000. 164(7):3913-3918. [DOD]
- Verdaguer J, Schmidt D, Amrani A, Anderson B, Averill N, Santamaria P. Spontaneous autoimmune diabetes in monoclonal T cell nonobese diabetic mice. J Exp Med 1997. 186(10):1663-1676. [DOD] [CrossRef]
- Wong FS, Visintin I, Wen L, Flavell RA, Janeway CA Jr. CD8 T cell clones from young nonobese diabetic (NOD) islets can transfer rapid onset of diabetes in NOD mice in the absence of CD4 cells. J Exp Med 1996. 183(1):67-76. [DOD] [CrossRef]
- Kurts C, Carbone FR, Barnden M, Blanas E, Allison J, Heath WR, Miller JF. CD4+ T cell help impairs CD8+ T cell deletion induced by cross-presentation of self-antigens and favors autoimmunity. J Exp Med 1997. 186(12):2057-2062. [DOD] [CrossRef]
- Herold KC, Hagopian W, Auger JA, Poumian-Ruiz E, Taylor L, Donaldson D, Gitelman SE, Harlan DM, Xu D, Zivin RA, Bluestone JA. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med 2002. 346(22):1692-1698. [DOD] [CrossRef]
- Keymeulen B, Vandemeulebroucke E, Ziegler AG, Mathieu C, Kaufman L, Hale G, Gorus F, Goldman M, Walter M, Candon S, et al. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med 2005. 352(25):2598-2608. [DOD] [CrossRef]
- Cooke A, Phillips JM, Parish NM. Tolerogenic strategies to halt or prevent type 1 diabetes. Nat Immunol 2001. 2:810-815. [DOD] [CrossRef]
- Sherry NA, Kushner JA, Glandt M, Kitamura T, Brillantes AM, Herold KC. Effects of autoimmunity and immune therapy on beta-cell turnover in type 1 diabetes. Diabetes 2006. 55(12):3238-3245. [DOD] [CrossRef]
- Sherry NA, Chen W, Kushner JA, Glandt M, Tang Q, Tsai S, Santamaria P, Bluestone JA, Brillantes AM, Herold KC. Exendin-4 improves reversal of diabetes in NOD mice treated with anti-CD3 monoclonal antibody by enhancing recovery of beta-cells. Endocrinology 2007. 148(11):5136-5144. [DOD] [CrossRef]
- Cobbold SP, Castejon R, Adams E, Zelenika D, Graca L, Humm S, Waldmann H. Induction of foxP3+ regulatory T cells in the periphery of T cell receptor transgenic mice tolerized to transplants. J Immunol 2004. 172(10):6003-6010. [DOD]
- You S, Leforban B, Garcia C, Bach JF, Bluestone JA, Chatenoud L. Adaptive TGF-beta-dependent regulatory T cells control autoimmune diabetes and are a privileged target of anti-CD3 antibody treatment. Proc Natl Acad Sci U S A 2007. 104(15):6335-6340. [DOD] [CrossRef]
This article has been cited by other articles:

|
Pathogenic Mechanisms in Type 1 Diabetes: The Islet is Both Target and Driver of Disease
Graham KL, Sutherland RM, Mannering SI, Zhao Y, Chee J, Krishnamurthy B, Thomas HE, Lew AM, Kay TW
Rev Diabet Stud 2012. 9(4):148-168
|
|

|
CNTF protects MIN6 cells against apoptosis induced by Alloxan and IL-1beta through downregulation of the AMPK pathway
Santos GJ, Oliveira CA, Boschero AC, Rezende LF
Cell Signal 2011. 23(10):1669-1676
|
|

|
Autoimmunity and inflammation: murine models and translational studies
Hall SW, Cooke A
Mamm Genome 2011. 22(7-8):377-389
|
|

|
B cell depletion in autoimmune diabetes: insights from murine models
Chamberlain JL, Attridge K, Wang CJ, Ryan GA, Walker LS
Expert Opin Ther Targets 2011. 15(6):703-714
|
|

|
The role of natural killer T (NKT) cells in the pathogenesis of type 1 diabetes
Gomez-Diaz RA, Aguilar MV, Meguro EN, Marquez RH, Magana EG, Martinez-García MC, Rodarte NW, Aguilar-Salinas CA, Canche-Pool E, Ortiz-Navarrete V
Curr Diabetes Rev 2011. 7(4):278-283
|
|

|
What causes type 1 diabetes? Lessons from animal models
Buschard K
APMIS Suppl 2011. 119(s132):1-19
|
|

|
Immune cell crosstalk in type 1 diabetes
Lehuen A, Diana J, Zaccone P, Cooke A
Nat Rev Immunol 2010. 10(7):501-513
|
|

|
Roles for TGF-beta and programmed cell death 1 ligand 1 in regulatory T cell expansion and diabetes suppression by zymosan in nonobese diabetic mice
Burton OT, Zaccone P, Phillips JM, De La Pena H, Fehervari Z, Azuma M, Gibbs S, Stockinger B, Cooke A
J Immunol 2010. 185(5):2754-2762
|
|

|
Harnessing CD8+ regulatory T cells: therapy for type 1 diabetes?
Zaccone P, Cooke A
Immunity 2010. 32(4):504-506
|
|

|
Breakthrough in diabetes therapy ... Just around the corner?
Rudert WA, Trucco M
Rev Diabet Stud 2009. 6(2):76-80
|
|
|


)
)
)
)

