Editorial
| Rev Diabet Stud,
2007,
4(1):6-12 |
DOI 10.1900/RDS.2007.4.6 |
Cystic Fibrosis-Related Diabetes in Adults: Where Can We Go From Here?
Harold W. de Valk1, Eduard A. van der Graaf2
1Department of Internal Medicine, University Medical Centre Utrecht, Heidelberglaan 100, 3584 CX Utrecht, The Netherlands.
2Department of Respiratory Medicine, University Medical Centre Utrecht, Heidelberglaan 100, 3584 CX Utrecht, The Netherlands.
Address correspondence to: Harold W. de Valk, e-mail: h.w.devalk@umcutrecht.nl
Keywords: CFRD, pulmonary function, diabetic complications, continuous glucose monitoring, lung transplantation, pregnancy
Abstract
Cystic fibrosis (CF), a dysfunction of the exocrine glands, is one of the most frequently diagnosed genetic diseases. It is characterized by chronic pulmonary disease and pancreatic deficiency. Cystic fibrosis-related diabetes (CFRD) is a complication of CF and develops from impaired glucose tolerance via postprandial hyperglycemia with fasting normoglycemia to full-blown diabetes with fasting and postprandial hyperglycemia. CFRD is related to decreased life expectancy, most notably in female patients, as well as to decreased pulmonary function and body weight reduction, which can be improved with adequate insulin therapy. Insulin therapy is accepted in full-blown diabetes but the treatment required by lesser degrees of abnormal glucose metabolism is unknown and needs to be clarified. Chronic organ complications of diabetes are seen only in full-blown diabetes with a particular tendency to affect the autonomous nervous system. Continuous glucose measurement techniques have opened new fields of investigation, particularly in relation to CF-related complications. Insulin therapy needs to be intensified and insulin pump therapy should receive more attention. While improvements in therapy, including lung transplantation, have resulted in increased life expectancies, other issues, such as fertility problems and pregnancy, have raised new questions. All of these need to be addressed to find new treatment options for CFRD patients. In this article we aim to illustrate how these new questions in the treatment of adult patients with CFRD could be answered.
Introduction
Cystic fibrosis (CF) is one of the most common inborn errors of metabolism, occurring in one in every 3000 live births in Caucasian populations but at a much lower frequency in other ethnic groups [1]. This means that the incidence of CF is six times higher than that of phenylketonuria. In many, but certainly not all patients, the lungs are primarily affected. Table 1 shows the many different potential clinical manifestations and problems associated with CF. Some patients are diagnosed soon after birth while in others the diagnosis is made only after the birth of an affected sibling or in adulthood.
Table
1.
Manifestations of cystic fibrosis |
|
 |
 |
The basic defect in CF is an abnormal transmembrane product in epithelia, the 'cystic fibrosis transmembrane conductance regulator' (CFTR) [2]. Depending on the gene mutation, the protein does not reach the cell surface because of abnormal processing or does reach the cell surface but functions abnormally [1]. As a consequence, electrolyte and fluid handling is abnormal, leading to abnormal epithelial secretions. In the lung, there is thickened and sticky mucus associated with insufficient clearing, setting the stage for recurrent infections with dire consequences. In the intestine, desiccated secretions can lead to pancreatic, biliary and gall bladder problems.
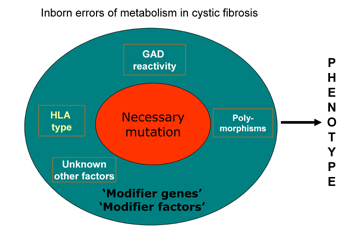
There is no clear genotype-phenotype relation. The most common mutation is the ΔF508 mutation, but approximately one thousand mutations have been described. The wide variety of clinical manifestations in individuals with this mutation and also within families is probably explained by modifier genes (Figure 1). These modifier genes can alter clinical expression through differences in the other relevant biochemical pathways implicated in the pathogenesis of organ manifestations of CF. According to this view, there are basically no monogenetic disorders, i.e. phenotypic manifestations of the disease are influenced by other genes and potentially by environmental factors. The detection of CF is principally based on clinical symptomatology combined with the demonstration of increased sweat chloride concentration. The treatment of CF is only supportive, but improvements in pulmonary, gastro-intestinal and endocrine care, as well as better patient participation, have resulted in a dramatic improvement of life expectancy, which is now around 40-50 years [3]. With longer life, new disease-related problems became evident, which were considered less relevant in the past.
 |
|
Figure 1. The pathogenesis of inherited monogenetic diseases. The figure shows the relation between the classic monogenetic mutation, modifying genes and other factors. Inborn errors are evoked by different genes, polymorphisms and other not yet fully known factors, which modify the metabolism of cystic fibrosis patients. |
|
CFRD is a prime example of such a complication. In this paper we discuss the epidemiology and pathogenesis, clinical manifestations and consequences and finally treatment options in connection with this complication. Attention will also be given to lung transplantations and pregnancy issues.
Cystic-fibrosis related diabetes mellitus
The biochemical defect present in CF also has an effect on the exocrine and endocrine pancreas. Patients develop exocrine pancreas insufficiency. In a series of patients, this is followed by endocrine insufficiency caused by destruction of the islets of Langerhans with ongoing fibrosis of the pancreas. Before long, insulin secretion diminishes and patients develop impaired glucose tolerance and then finally diabetes. The disease progresses from impaired glucose tolerance followed by a phase of increasing fasting glucose and finally becomes full-blown diabetes. Basically, this sequence of events in CFRD corresponds to the development of primary diabetes but at a slower pace and some authors suggest a classification based on the sequence. Impaired glucose tolerance is the first phase with normal fasting glucose combined with post-glucose load or postprandial values ranging form 7.8 to 11.0 mmol/l. Subsequently, CFRD develops without fasting hyperglycemia but with post-load postprandial values of 11.1 mmol/l or more. Finally, full-blown CFRD ensues with fasting hyperglycemia.
A number of questions arise in relation to the development of CFRD. Firstly, what is the clinical relevance of CFRD in these patients? Secondly, what is the risk of chronic organ complications in diabetes? Thirdly, if diabetes is especially relevant, how do we organize early detection and monitoring and, finally, what would be the best treatment options?
Specific consequences of CFRD
Mortality is an important parameter and a recent study by Milla et al. has shown that CFRD reduces life expectancy in patients with CF [3]. However, this effect was only seen in women and not in men. It may be the case that such a gender effect was only discovered because the application of modern treatment strategies increased life expectancy. Reasons cited for this gender difference include a general gender-associated effect on pulmonary function and consequently on respiratory function as well as gender-associated differences in lean body mass [4]. Clearly, these results point to the relevance of diabetes in cystic fibrosis, especially for women and these data concern mortality. The morbidity and psychological burden of the disease are further critical issues.
In CF, diabetes has been linked to an increased deterioration in pulmonary function and to problems in weight management. In addition, a number of studies have investigated the relationship between glycemic control and the development of respiratory problems, as outlined in the next sections.
Respiratory function
The presence of diabetes is particularly associated with a lower pulmonary function (FEV1) compared with patients without diabetes [5]. Also, retrospective studies have shown that pulmonary function declines faster in the years preceding diagnosis in patients with CFRD compared with those without CFRD, but is restored with insulin treatment [6-9]. It has been observed that hyperglycemia has detrimental effects on pulmonary structure and insulin deficiency on respiratory muscle strength. An increased susceptibility to infections has been put forward to explain this phenomenon [10].
Weight management
Hyperglycemia-associated loss of body weight is a common phenomenon. In the presence of hyperglycemia, it is difficult to redress the catabolic state and improve body weight. Insulin therapy is necessary to improve body weight in such a condition but may take months to years [11].
Necessity for glycemic control in CFRD
Considering the high rate of mortality and morbidity in CFRD, there is a clear need for optimal glycemic control beginning when the disease is diagnosed. However, the target levels for glycemic control remain unknown, i.e. we do not know if they are different from the usual targets for the prevention of chronic organ complications in diabetes. It may be the case that even more strict targets are needed but data elucidating this issue are not yet available. This question is reminiscent of the situation in diabetic pregnancy where lower glucose levels compared with normal diabetes are used, since pregnancy-related end points (embryonic, fetal and maternal) are differentially related to glucose control rather than chronic organ complications. The question of whether patients with CF, impaired fasting glucose (IFG) and impaired glucose tolerance (IGT) should be treated with similar intensity, is related to this issue.
Chronic organ complications in CFRD
The second question concerns the chronic organ complications expected in patients with CFRD. In a small group of patients, Andersen et al. recently showed that retinopathy occurred in 21% of non-transplanted patients and micro-albuminuria in 13% [12]. Neuropathy was not encountered. A higher prevalence of retinopathy, nephropathy, hypertension and neuropathy occurred after transplantation, but the numbers were very small. Very recently, Schwarzenberg et al. found that microvascular complications occur only with CFRD and fasting hyperglycemia; the first signs occurred after 5 years of disease duration [13]. In both studies, lung transplantation with its attendant medication distorts this picture and no reliable data exist from patients with or without transplantation divided into separate groups. Most problems seem to occur after transplantation, which may also be linked to the application of immunosuppressive agents. Although only a limited number of eligible patients participated in the Schwarzenberg study (59/192, 31%), autonomic neuropathy may be a prevalent finding in all forms of CFRD [13]. Macrovascular disease has not been reported so far [12, 13]. These early data mean that more needs to be learned about the development of chronic complications with the emphasis on neuropathy and the specific role of transplantation.
Glucose monitoring
The third critical issue involves detecting an abnormal glucose condition and monitoring. Repeated assessment of glucose levels is indicated because diabetes is associated with an increased risk of accelerated pulmonary decline and because many patients have silent hyperglycemia. From a practical point of view, annual assessment is a good option. In our clinic we prefer the annual oral glucose tolerance test because we want to detect any abnormalities. Patients with full-blown diabetes are referred to the endocrinology unit. Patients with IFG or IGT are referred as well, but initially they are not treated but closely monitored with regular glucose profiles, the emphasis being on the postprandial values since they are usually the first to increase. Treatment is withheld until full-blown diabetes has developed.
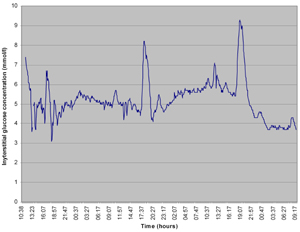
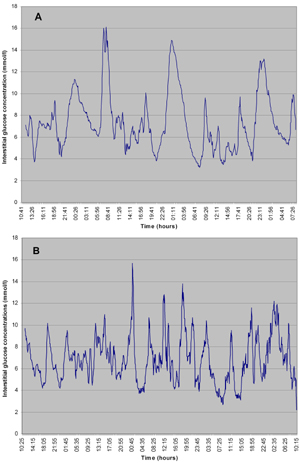
Continuous glucose monitoring (CGM) has expanded the scope of glucose monitoring, in the field of CF also. When we perform CGM in patients with IFG of IGT, we generally encounter postprandial hyperglycemia. This postprandial hyperglycemia is often related to food intake (Figure 2). With the advent of diabetes, this picture gradually evolves to postprandial and fasting hyperglycemia. Figure 2 shows data from our clinic. During these observations we often encounter a specific problem in CF related to nutrition. Patients are advised to sustain or improve body weight through repetitive small meals or, if this is not possible, they are temporarily given tube feeding. Figure 3 shows an example of the effects of repetitive small meals and snacks in advanced diabetes (without much residual beta-cell function). These conditions also illustrate the fact that glucose-lowering treatment has to be adjusted to the individual situation.
 |
|
Figure 2. Continuous glucose measurements in patients with glucose intolerance. Data are collected by continuous glucose sensor. Single small meal-related peaks are seen, the apex is below 11.0 mmo/l. |
|
 |
|
Figure 3. Continuous glucose measurements in patients with early CFRD. Data are collected by continuous glucose sensor. In both patients (A and B) peaks are mealtime-related, snack-related or related to additional ingestion. |
|
Treatment of CFRD
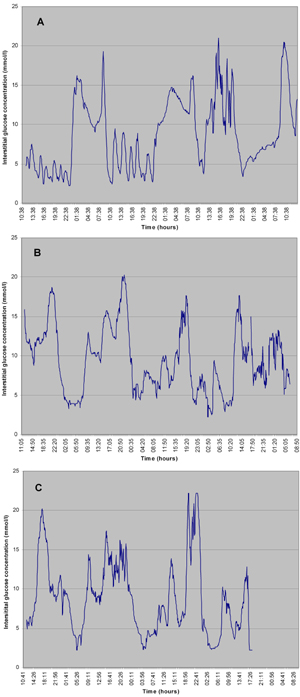
The observations from glucose monitoring in CFRD patients (Figure 4) brings us to the fourth issue, that of treatment. Nutritional treatment is obviously the first step and may be sufficient in early stages (IFG and IGT). The essential nutritional approach aims to prevent large episodic carbohydrate intake, especially of fluid. This is sometimes difficult since many patients favor these liquid energy providers. Before long, severe peaks of postprandial glucose appear. They are clinically silent but pathogenetically important for the development of organ complications and they reflect an increased risk of decline in pulmonary function. These observations show that CGM may detect relevant peaks that are missed by episodic fingerstick measurements and underscore the importance of adequate postprandial measurements.
 |
|
Figure 4. Continuous glucose measurements in full-blown CFRD. Glycemic peaks in all three pateints (A, B and C) are meal-time-related, snack-related or related to additional nutritional compounds. The peaks are higher and variability is higher than in early CFRD. |
|
When pharmacological treatment is indicated, insulin therapy is the first choice. Sulphonylurea derivatives act by increasing insulin secretion, but the progressive destruction of the beta-cells shows that these agents have limited value in CFRD, certainly in the longer term. Metformin acts by decreasing hepatic glucose output and is not a good option in view of the postprandial abnormalities. Thiazolidinedione-derivatives decrease insulin resistance but insulin resistance plays a minor role in the pathogenesis of CFRD. Therefore, insulin is the best choice although randomized trials have not been performed. On the basis of the glucose profiles, we can generally start with short-acting insulin with the meals and, if necessary and possible, with the snacks in-between as well. This latter course means that insulin analogues with rapid- and short-acting profiles are the insulins of choice. With fasting hyperglycemia, a long-acting insulin should be added. If very variable glucose profiles are observed, continuous subcutaneous insulin infusion with an externally-worn insulin pump is a good option. There is no general reason why this method can not be used in CFRD or why it should have worse results than those obtained in patients with other forms of diabetes.
As mentioned before, there are no data available on specific target levels for plasma glucose values in CFRD. The intuitive preference is for glucose profiles to be as normal as possible with fasting levels below 5.5 mmol/l and postprandial levels below 7.8 mmol/l. Attempts to achieve optimal control are limited by hypoglycemia, which is an ever-present danger during glucose-lowering therapy and also in CFRD. Because HbA1c levels are a rough parameter that does not always reflect glucose variabilities and the number of hyperglycemic peaks, fingerstick glucose measurements are equally important. In selected cases, CGM may help.
In summary, poor glucose control is associated with an increased rate of pulmonary function loss and weight loss and it seems reasonable to suggest that optimal control will be beneficial to both. Insulin is the therapy of choice in patients with full-blown diabetes and intensive therapy is required, together with nutritional support. In patients with IFG or IGT, nutritional support is indicated. There is no data on the beneficial effect of pharmacological treatment.
CFRD and pregnancy
With the prolongation of life expectancy, the issue of pregnancy appears to be more critical. Most men with CF are infertile because of the absence or obliteration of the vas deferens. Women have a reduced fertility caused by changes in the cervical mucus, slowed transport of the oocyte through the Fallopian tubes or blockage of these tubes. Besides these specific reasons, other general factors can also play a role in pregnancy with CFRD.
When a woman with CF does not become pregnant with a normal fertile partner, assisted conception is the next step. However, it is advisable to test the male partner before starting assisted conception to ensure that male infertility or subfertility does not undermine preparations for what is, at best, a low chance of pregnancy success. If the male partner is fertile, artificial insemination or in vitro fertilization can be considered. Of course, a thorough examination of the woman's overall physical condition should be carried out and pulmonary reserve is a very important factor in assessing whether pregnancy is a reasonable option.
If assisted conception is scheduled, then potential teratogenic medication should be stopped. The diabetes associated with CF imposes an additional problem. Meticulous glycemic control is mandatory, as it is in women with type 1 or type 2 preconceptional diabetes [14-18]. In the case of reduced life expectancy and assisted conception in particular, the future of the child should be discussed if the mother should become severely handicapped or die. It is important to clarify who takes care of the child and whether there is potentially sufficient family and peer support. In selected cases, non-commercial oocyte donation can be an option even though the associated difficulties and ethical issues must be considered. Certainly, these topics will be the focus of more attention in the coming years.
Lung transplantation
When, despite all efforts to improve pulmonary function, respiratory failure occurs, lung transplantation is the only option left. The prognosis with single or double lung transplantation is quite good in patients with CF and life expectancy of graft and patient is steadily increasing. Normally, transplanted patients start a second life and are over the moon. However, they have to take life-long immunosuppressive agents with their associated toxicities. In this scenario, prednisone, as an anti-inflammatory agent, is associated with an increase in insulin resistance. The calcineurin inhibitor tacrolimus is particularly associated with post-transplant diabetes mellitus induced by interfering beta-cell function. This means that some patients with CF develop diabetes after transplantation as a consequence of tacrolimus medication and not because of CF. Of course, both factors may contribute to the development of diabetes in these patients.
The long term risks of lung transplantation in non-CF recipients also threaten CF-recipients, such as acute or chronic rejection and bronchiolitis obliterans. After a successful lung transplant, questions about pregnancy may start to arise. Pregnancy is complicated by concerns about the effect of pregnancy on respiratory function, the teratogenic effects of certain immunosuppressive agents, the negative effects on immune status of changing immunosuppressive agents, other medications and the above-mentioned ethical issues.
Summing up
Diabetes is a major complication in patients with CF, probably related to an increased loss of pulmonary function, problems in maintaining weight and, in female patients in particular, increased mortality. Regular testing is mandatory as is the prompt treatment of any diabetes detected. If diabetes develops in CF patients, intensive insulin therapy is required, including insulin pump therapy. Therapy of milder glucose abnormalities (IFG, IGT), besides nutritional intervention, is yet of unknown benefit. Pregnancy is an option in specific circumstances, as mentioned above, but diabetes has to be strictly regulated, teratogenic medication stopped and pulmonary function maintained. This is true a fortiori in the case of lung transplantation. To improve the health of CFRD patients, further research efforts are needed, especially in the field of early glucose abnormalities, pregnancy issues and transplantation.
References
- Boucher RC. Cystic fibrosis. In: Harrison’s principals of internal medicine. 2005, 16th edition. McGraw-Hill Medical Publishing Company; chapter 241, pp. 1543-1546. [DOD]
- Ratjen F, Doring G. Cystic fibrosis. The Lancet 2003. 361(9358):681-689. [DOD] [CrossRef]
- Milla CE, Billings J, Moran A. Diabetes is associated with dramatically decreased survival in female but not male subjects with cystic fibrosis. Diabetes Care 2005. 28:2141-2144. [DOD] [CrossRef]
- Davis PB. The gender gap in cystic fibrosis survival. J Gend Specif Med 1999. 2(2):47-51. [DOD]
- Koch C, Rainisio M, Madessani U, Harms HK, Hodson ME, Mastella G, McKenzie SG, Navarro J, Strandvik B. Presence of cystic fibrosis-related diabetes mellitus is tightly linked to poor lung function in patients with cystic fibrosis: data from the European Epidemiologic Registry of Cystic Fibrosis. Pediatr Pulmonol 2001. 32(5):343-350. [DOD] [CrossRef]
- Finkelstein SM, Wielinski CL, Elliott GR, Warwick WJ, Barbosa J, Wu SC, Klein DJ. Diabetes mellitus associated with cystic fibrosis. J Pediatr 1988. 112(3):373-377. [DOD] [CrossRef]
- Lanng S, Thorsteinsson B, Nerup J, Koch C. Influence of the development of diabetes mellitus on clinical status in patients with cystic fibrosis. Eur J Pediatr 1992. 151:684-687. [DOD] [CrossRef]
- Milla CE, Warwick WJ, Moran A. Trends in pulmonary function in patients with cystic fibrosis correlate with the degree of glucose intolerance at baseline. Am J Resp Crit Care Med 2000. 162:891-895. [DOD]
- Wilson DC, Stewart C, Hanna A. Cystic fibrosis-related diabetes in adults: clinical implications. Pulmonology 1996. 13:323. [DOD]
- Brennan AL, Geddes DM, Gyi KM, Baker EH. Clinical importance of cystic fibrosis-related diabetes. J Cyst Fibros 2004. 3(4):209-222. [DOD] [CrossRef]
- Mackie AD, Thornton SJ, Edenborough FP. Cystic fibrosis-related diabetes. Diabet Med 2003. 20(6):425-436. [DOD] [CrossRef]
- Andersen HU, Lanng S, Pressler T, Laugesen CS, Mathiesen ER. Cystic fibrosis-related diabetes. Diabetes Care 2006. 29:2660-2663. [DOD] [CrossRef]
- Schwarzenberg SJ, Thomas W, Olsen TW, Grover T, Walk D, Milla C, Moran A. Microvascular complications in cystic fibrosis-related diabetes. Diabetes Care 2007. 30(5):1056-1061. [DOD] [CrossRef]
- Evers IM, de Valk HW, Visser GH. Type 1 diabetic pregnancies are still at high risk for complications: outcome of a nationwide study. BMJ 2004. 328:908-915. [DOD] [CrossRef]
- Hawthorne G, Robson S, Ryall EA, Sen D, Roberts SH, Ward Platt MP. Prospective population based survey of outcome in pregnancy in diabetic women: results of the Northern Diabetic Pregnancy Audit, 1994. BMJ 1997. 315(7103):279-281. [DOD]
- Casson IF, Clarke CA, Howard CV, McKendrick O, Pennycook S, Pharoah PO, Platt MJ, Stanisstreet M, van Velszen D, Walkinshaw S. Outcomes of pregnancy in insulin dependent diabetic women: results of a five year population cohort study. BMJ 1997. 315(7103):275-278. [DOD]
- Clausen TD, Mathiesen E, Ekbom P, Hellmuth E, Mandrup-Poulsen T, Damm P. Poor pregnancy outcome in women with type 2 diabetes. Diabetes Care 2005. 28(2):323-328. [DOD] [CrossRef]
- De Valk HW, van Nieuwaal NH, Visser GH. Pregnancy outcome in type 2 diabetes mellitus: a retrospective analysis from the Netherlands. Rev Diabet Stud 2006. 3(3):134-142. [DOD] [CrossRef]
This article has been cited by other articles:
|


)
)
)
)
)



