Congenital and Environmental Factors Associated with Adipocyte Dysregulation as Defects of Insulin Resistance
Introduction
The metabolic syndrome refers to the cluster of several cardio-metabolic risk factors, including central obesity, hyperglycemia, dyslipidemia (elevated triglycerides, low levels of high density lipoprotein cholesterol) and elevated blood pressure, all of which have been linked to insulin resistance [1]. The metabolic syndrome is a common metabolic disorder resulting from the increased prevalence of obesity. The disorder is defined in various ways and different definitions require different cut points and have different mandatory inclusion criteria [2]. The apparent pathophysiology is insulin resistance with excessive flux of fatty acids. Insulin resistance was brought to the fore by Gerald Reaven in 1988 [3]. Reaven referred to the disorder as Syndrome X, but it is also commonly known as Reaven’s syndrome, insulin resistance syndrome and metabolic syndrome. The latter is the most widely used terminology.
The first operational definition of the metabolic syndrome proposed by the World Health Organization [4] was hyperglycemia and/or insulin resistance as a central feature associated with two or more related metabolic abnormalities. The metabolic syndrome has also been defined by readily assessable clinical and biochemical measures to improve identification of at-risk subjects [5, 6]. Large epidemiological surveys show that the metabolic syndrome, according to most definitions, is common [7]. The prevalence of the metabolic syndrome increases with age and varies with gender and ethnicity [8]. Prospective studies have established that the metabolic syndrome is associated with a two-fold increased risk in cardiovascular disease [9]. Proposed pathophysiological explanations for the metabolic syndrome include insulin resistance, chronic inflammation and ectopic fat accumulation following adipose tissue saturation with fatty acids [10]. However, the central core of the metabolic syndrome may be failed evolutionary adaptation of adipose tissue to affluent lifestyles and excess energy intake.
Adipose tissue in human evolution
Adipose tissue is traditionally viewed as a site of excess energy stored as triglycerides (TGs). When needed elsewhere in the body, that energy is released as fatty acids (FAs) [11-13]. Although often cited in human evolutionary theory, the role of the adipose tissue in human evolution has not been clearly elucidated. As adipose tissue is not fossilized, clinical studies have relied on comparative data [13]. Nowadays, fat depots of normal persons contain sufficient energy to survive one month or more of total starvation. This excess energy storage is less important now since food supplies are plentiful. In previous eras of human history, long periods of famine after years of bad harvests and long winters claimed many lives [14]. Those who survived despite limited external energy supplies were those with large fat deposits. Genetic selection favored those who could accumulate sufficient excess fat to survive long periods of starvation [15]. Such conditions may have caused selection of a so called "thrifty gene", which, in the present era of abundant food supplies, exacerbates obesity [16, 17]. This selection of obesity genes by evolution (congenital factor) and overnutrition due to an affluent life style (environmental factor) may be the leading causes of severe obesity worldwide and particularly in third world countries [18]. The metabolic syndrome was thus introduced into modern civilization (Figure {f1}).
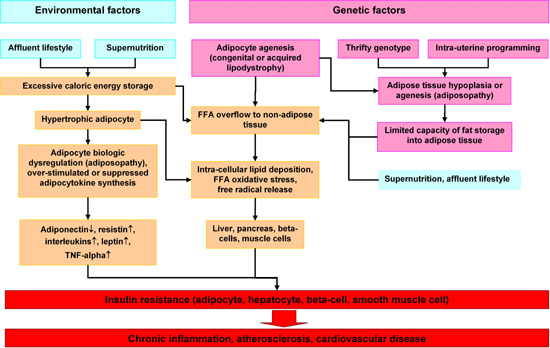 |
 |
Figure 1. Hypothetical program of adipocyte dysregulation (adiposopathy) in the pathogenesis of the insulin resistance syndrome. Environmental and genetic factors may work in concert to cause dysregulation of hypertrophic adipocytes, FFA overflow in non-adipose tissue such as liver and pancreas and finally insulin resistance and chronic diseases. |
|
Thrifty genotype and phenotype hypothesis
A rapidly growing epidemic of type 2 diabetes and cardiovascular disease has afflicted developing countries throughout Southeast Asia [19]. By 2025, India is predicted to have >60 million diabetic patients, and coronary heart disease (CHD) is expected to be the leading cause of adult mortality [20, 21]. In other words, one in five diabetic patients worldwide are expected be Indian, and three in four from developing countries [21]. This phenomenal rise in diabetes and CHD has been ascribed to rapid demographic, nutritional and socioeconomic changes, the so-called epidemiological, nutritional and economic transition [22].
The thrifty genotype and the thrifty phenotype are two nonexclusive explanations for the insulin resistance epidemic in developing countries [16, 23-25]. Asian populations have faced undernutrition for many generations; however, the diabetes epidemic is of recent origin [19]. Susceptibility to diabetes is ascribed to the evolution of thrifty genes, which enhanced survival in the past when food supplies were scarce and intermittent but which have become physically detrimental in contemporary conditions of plentiful food and sedentary lifestyles [23-26].
The evolutionary advantage of Homo sapiens lay in the way he obtained food, which in turn depended on physical activity. A consistent food supply, however, could never be relied upon. Certain genes evolved to regulate the efficient intake and utilization of fuel stores, thus ensuring survival during famine. These genes were called "thrifty genes" in 1962 [16]. In addition there is evidence that the human genome has remained essentially unchanged for as long as 10,000 years and certainly over the past 40-100 years [26, 27]. Although the absolute caloric intake of modern humans is probably lower than that of early hunter-gatherers, the caloric balance in modern adult populations is nevertheless positive because of the sedentary lifestyle in the today’s society [17, 26]. The combination of an abundant and reliable food supply and lack of physical activity eliminates the biochemical cycles which evolved from feast-famine and physical activity-rest cycles. Hence, certain metabolic processes no longer occur in cycles, which ultimately lead to metabolic disorders such as obesity and type 2 diabetes [17, 26].
An alternative explanation is the recently proposed thrifty phenotype hypothesis which ascribes the obesity epidemic to an unfavorable intrauterine environment [23, 24, 27]. The crucial importance of the intrauterine period in determining health and disease is understandable given that humans begin life as a single cell and that more than three quarters of total cell divisions occur in utero [28, 29]. Substantial growth and development is completed before birth, as demonstrated by the inverse relationship between birth weight and risk of diabetes and metabolic syndrome later in life [30-32]. The thrifty phenotype hypothesis proposes that early-life metabolic adaptations promote survival, with the developing organism responding to environmental cues by selecting an appropriate growth trajectory [33, 34].
The two explanations are not necessarily mutually exclusive and may be complimentary. It has been said that the rapidly emerging epidemic of the metabolic syndrome in the twentieth Century reflects the rapidly accelerating pace of niche construction relative to the slower physiological combination of developmental plasticity and parental effects [17].
Environmental factors - overnutrition, obesity and adipocyte hypertrophy
Adipose tissue becomes important when increased energy storage is required. However, these deposits are fairly limited. Although the excess energy may be efficiently stored, fat storage capacity is limited [35, 36]. The varying functional capacity of adipose tissue explains the incomplete overlapping between the metabolic syndrome and obesity. Obesity is characterized by elevated fat accumulation caused by enlarged adipocytes [37-39]. The typical adipocyte without accumulated fat is approximately 10-12 micrometers in diameter, the approximate size of a lymphocyte. After normal fat accumulation, the diameter increases 10-fold, a 1000-fold volume increase [39].
Researchers have demonstrated associations between adipocyte enlargement and metabolic change, e.g. lipid metabolism, as well as the release of cytokines, chemokines and many other biologically active molecules, commonly known as adipokines [40, 41]. These secreted products may participate in developing a chronic low-grade inflammatory state which is "common soil" for the pathogenesis of metabolic and cardiovascular complications of obesity [35, 42-46].
The effects of ectopic fat deposition and the overflow of lipids into muscles and liver on the function of beta-cells
For the first time in history, large human populations have a readily available, inexpensive and palatable food supply. This has created a situation, on a scale not previously encountered, in which the capacity to store triglycerides in adipocytes has become an important determinant of human health. An inadequate number of adipocytes (e.g., lipodystrophia) or overcapacity of adipocytes (e.g., severe obesity), produce similar and unfavorable metabolic effects where ectopic triglyceride stores may appear in liver, muscle and pancreatic islet tissue [47, 48].
Clearly, under normal conditions, the storage and release of TG and FA, respectively, are both coordinated and tightly regulated such that lipid fuels are stored during the immediate postprandial periods and released during periods of fasting [49]. More recent studies have demonstrated that impaired regulation of storage and release of energy by adipose tissue elevate plasma FA levels, in particular when the release of FA becomes dissociated from energy requirements in other organs and tissues. Excessive metabolism of FA, including storage of TG, occurs in nonadipose tissues [47, 50]. The expanded view of ectopic metabolism or accumulation of FA or TG is that it causes dysfunction, or lipotoxicity, in these organs and tissues [51-54].
Clinical evidence indicates that overflow to non-adipose tissue results from the limited storage capacity of congenital adipocytes, either from intra-uterine programming or excess energy intake due to an affluent life style and overeating, which can trigger the process of insulin resistance. Firstly, the failure to develop adequate adipose tissue mass (also known as 'lipodystrophy') causes severe insulin resistance and diabetes [55] due to ectopic storage of lipids in the liver, skeletal muscle and pancreatic insulin-secreting beta-cell [56, 57].
Secondly, most obese patients also shunt lipids into skeletal muscle, the liver and apparently into beta-cells [58-60]. The importance of this finding is exemplified by several studies demonstrating that the degree of lipid infiltration into skeletal muscle and the liver highly correlates with insulin resistance [61].
The role of increased fatty acid flux in muscle and insulin resistance
Excess FA trapping in muscles combined with reduced FA oxidation increases TG synthesis. Indeed, increased intramyocellular TG correlates strongly with insulin resistance [52, 54, 61]. Although some studies suggest that intramyocellular TG is a lipotoxic substance in muscle tissue, it more likely functions only as a marker of reduced oxidation relative to FA availability [62]. Of interest in this regard are findings that insulin-resistant and diabetic individuals have a decreased fractional FA uptake and, in some cases, absolute FA uptake by muscles has been observed [52, 63]. However, normal or only slightly reduced plasma FA levels during the immediate postprandial period in insulin-resistant individuals, when plasma FA levels should be dramatically suppressed, combined with modestly reduced FA extraction by the muscle, can still cause excess FA in muscle [64]. Additionally, considerable evidence demonstrates that insulin-resistant individuals exhibit defective mitochondrial oxidation of FA [52, 54], which is reversible by weight loss and exercise [65].
The role of increased fatty acid flux to beta-cells and defects in insulin secretion
Plasma FA can be taken up by pancreatic beta-cells where they can stimulate insulin secretion under certain conditions [66]. The latter apparently occurs because FAs generate long-chain fatty acyl CoA, which contributes to the synthesis of signaling molecules such as DAG and phosphatidic acid or directly stimulates PKC isoforms involved in insulin secretion [67]. Of particular relevance to insulin resistance is the finding that circulating FA are critical in the pancreatic response to glucose in the fasting state [68, 69]. Thus, the high basal insulin response characteristic of insulin resistance might be partly due to increased FA levels before administration. High-plasma FA uptake by beta-cells also increases FA storage as TG [67]. Clinical evidence clearly shows that continued high-cytosolic FA concentrations ultimately decrease insulin secretion [67]. Finally, animal models indicate that chronic exposure of beta-cells to high FA concentrations and glucose causes apoptosis in ZDF rats [51, 66, 70].
Non-alcoholic fatty liver disease - the role of increased fatty acid flux to hepatocytes
Non-alcoholic fatty liver disease (NAFLD) is an ectopic fat storage disorder strongly associated with insulin resistance [71, 72]. Patients with NAFLD are apparently predisposed to type 2 diabetes because of its close association with insulin resistance and its consequences, including hyperinsulinemia, glucose intolerance, hypertriglyceridemia, decreased high-density lipoprotein cholesterol concentration and hypertension [73]. Although most commonly associated with obesity, NAFLD may also be observed in non-obese subjects [74, 75].
Convincing evidence suggests that ectopic fat accumulation in insulin-sensitive tissues is associated with insulin resistance, independent of overall obesity. However, the precise causes and mechanisms underlying fat accumulation in skeletal muscle and the liver are unclear [76]. Identifying why some individuals store fat in insulin-sensitive tissues may be of great importance in developing new insulin-sensitizing agents and for optimizing current therapies [77, 78].
The published literature clearly demonstrates that dysregulated partitioning of FA between storage sites (adipocytes) and sites of utilization (muscle, liver and beta-cells) upsets lipid homeostasis in the latter tissues. Deleterious outcomes include TG accumulation and insulin resistance [47, 48, 57, 58]. This fact suggests that, in the metabolic syndrome, adipose tissue insulin resistance is the only cause of insulin resistance in other sites [79]. Thus, inadequate energy storage in adipocytes causes abnormal lipid metabolism and possibly insulin resistance, inflammation and cell death in alternative storage sites [80].
Lipodystrophy
Lipodystrophies are rare, inherited and acquired disorders characterized by the selective loss of adipose tissue [81]. The main forms can be classified according to their origin, either genetic or acquired, and subclassified by their clinical pattern [36, 55, 81]. Interestingly, the selective loss of adipose tissue is also frequently associated with marked insulin resistance and complications such as type 2 diabetes, dyslipidemia and hypertension [56, 82]. The final outcome of loss of physiological lipid partitioning is total lipodystrophy, a disorder characterized by the absence of adipose tissue [36]. Interestingly, patients with total lipodystrophy, who have no intra-abdominal fat or visceral fat, are severely insulin-resistant. Partial lipodystrophy patients may also exhibit ectopic fat deposition in several organs as well as visceral fat, hypertriglyceridemia and insulin resistance [82].
Despite marked phenotypic and genotypic heterogeneity, most lipodystrophic syndromes are predisposed to metabolic complications commonly observed in obese patients, such as insulin resistance, diabetes mellitus, hepatic steatosis and dyslipidemia [56]. Thus, normal levels of adipose tissue and normal fat distribution are apparently critical for optimizing regulation of lipid and energy metabolism [83]. The precise mechanisms by which the lack of adipose tissue causes hypertriglyceridemia remain unknown. Anecdotal kinetic studies in hyperglycemic patients with lipodystrophies reveal accelerated lipolysis and increased free fatty acid turnover, which drives hepatic triglyceride and very low-density lipoprotein synthesis. Other possible causes of dyslipidemia and ectopic triglyceride accumulation in the liver and skeletal muscles have been proposed but require further confirmation [81].
Adipocytokines
Although adipose tissue was considered inert tissue until 40 years ago, several vital functions have been associated with this highly active tissue [11, 35, 84]. Adipocytokines are bioactive mediators released from adipose tissue including adipocytes and other cells present in fat tissues [85-87]. These include several novel and highly active molecules abundantly released by adipocytes, such as leptin, resistin, adiponectin or visfatin as well as other well known cytokines possibly released by inflammatory cells infiltrating fat, such as TNF-alpha, IL-6, MCP-1 and IL-1 [41, 43]. Leptin and adiponectin are the most abundantly expressed adipocytokines within the adipose tissue [88-90].
These molecules, along with FA, significantly affect total body glucose, lipid metabolism and insulin sensitivity [85, 91, 92]. Adipocytokine release may elucidate the relationship between obesity and cardiovascular phenotypes, including hypertension and atherosclerosis, mainly through their ability to affect and modify endothelial and vascular function as well as by their modulating effects on immune functions, which have been elucidated in several reviews [91-94].
Psammomys obesus, an animal model of environmental-gene interaction of type 2 diabetes
Psammomys obesus, a rat found in semi-desert regions of North African and the Eastern Mediterranean, exhibits insulin resistance similar to that in human type 2 diabetes. In its native habitat, P. obesus feeds mainly on the low-calorie Atriplex halimus plant, and is neither obese nor hyperglycemic. However, when given calorie-rich rodent food feed ad libitum in captivity, it tends to become moderately obese and develops postprandial hyperglycemia [95-97].
The P. obesus model elucidates environmental and genetic factors in the pathogenesis of insulin resistance [95]. P. obesus is characterized by muscle insulin resistance and impaired insulin signaling on a high-energy diet. Insulin resistance imposes a vicious cycle of hyperglycemia and compensatory hyperinsulinemia, leading to beta-cell failure and increased proinsulin secretion [96].
Conclusions
In recent decades, the prevalence of insulin resistance and diabetes has increased in Western populations as well as in Asian populations adopting "Western" lifestyles. This trend indicates the impact of environmental factors such as diet, obesity and physical activity on the pathogenesis of the metabolic syndrome. According to the ‘thrifty gene’ hypothesis, a cluster of genetic defects, which originally provided some populations with a genetic advantage, could predispose some ethnic groups to insulin resistance and diabetes in the presence of an increased food supply. This concept has arisen from hypothesized interactions between genetic, intrauterine and environmental factors.
The inherited defects resulting from adipose tissue differentiation are not fully adapted to modern lifestyles and excess caloric intake. Inadequate adipose tissue mass and overnutrition cause adipocytes to increase fat cell size and stimulate release of insulin-resistant adipocytokines. These processes cause severe insulin resistance, endothelial dysfunction, a proinflammatory state and promote the progression of atherosclerosis. Overnutrition as excess FA overflow to ectopic fat storage organs, such as the liver, muscle, and pancreatic beta-cells, cause functional alternation in these cells and shift the intra-cellular metabolic pathway towards insulin resistance.
Cumulatively, these observations indicate that the 'adipocyte dysregulation-adiposopathy' hypothesis actually identifies congenital or environmental triggering factors as primary causes of insulin resistance.
|

)
