Review
| Rev Diabet Stud,
2009,
6(3):148-158 |
DOI 10.1900/RDS.2009.6.148 |
A Molecular Level Understanding of Zinc Activation of C-peptide and its Effects on Cellular Communication in the Bloodstream
Wathsala Medawala, Patrick McCahill, Adam Giebink, Jennifer Meyer, Chia-Jui Ku, Dana M. Spence
Department of Chemistry, Michigan State University, East Lansing, MI 48824, USA
Address correspondence to: Dana M. Spence, e-mail: dspence@chemistry.msu.edu
Manuscript submitted October 15, 2009; resubmitted October 26, 2009; accepted November 2, 2009.
Keywords: diabetes, C-peptide, red blood cell, glucose, zinc, Zn2+, Ca2+, signaling, platelets, nitric oxide, ATP
Abstract
Inspired by previous reports, our group has recently demonstrated that C-peptide exerts beneficial effects upon interactions with red blood cells (RBCs). These effects can be measured in RBCs obtained from animal models of both type 1 diabetes and type 2 diabetes, though to different extents. To date, the key metrics that have been measured involving C-peptide and RBCs include an increase in glucose uptake by these cells and a subsequent increase in adenosine triphosphate (ATP) release. Importantly, to date, our group has only been able to elicit these beneficial effects when the C-peptide is prepared in the presence of Zn2+. The C-peptide-induced release of ATP is of interest when considering that ATP is a purinergic signaling molecule known to stimulate the production of nitric oxide (NO) in the endothelium and in platelets. This NO production has been shown to participate in smooth muscle relaxation and subsequent vessel dilation. Furthermore, NO is a well-established platelet inhibitor. The objective of this review is to provide information pertaining to C-peptide activity on RBCs. Special attention is paid to the necessity of Zn2+ activation, and the origin of that activation in vivo. Finally, a mechanism is proposed that explains how C-peptide is exerting its effects on other cells in the bloodstream, particularly on endothelial cells and platelets, via its ability to stimulate the release of ATP from RBCs.
Abbreviations: Arg - arginine; ATP - adenosine triphosphate; BB/Wor - BioBreeding/Worcester (animal model of spontaneous autoimmune diabetes); BBZDR/Wor - BioBreeding Zucker Diabetic Rat/Worcester (animal model of spontaneously non-insulin-dependent diabetes); Ca2+- bivalent ionic calcium; 14C - carbon 14 (radioactive isotope of carbon); Cr3+ - trivalent ionic chromium; C-peptide - connecting peptide; EDTA - ethylenediaminetetraacetic acid; eNOS - endothelial nitric oxide synthase; Fe2+ - bivalent iron cation; Fe3+ - trivalent iron cation; GLUT1 - glucosetransporter 1; HPLC - high performance liquid chromatography; K+ - potassium ion; L-NAME - NG-nitro-L-arginine methyl ester (NO synthase inhibitor); Lys - lysine; Na+ - sodium ion; NO - nitric oxide; P2X - purinergic receptor (cation-permeable ligand gated ion channels that open in response to the binding of extracellular ATP); P2Y - purinergic receptor (stimulated by nucleotides such as ATP); PS - phosphatidylserine; RBC - red blood cell; Zn2+ - bivalent ionic zinc
C-peptide secretion from pancreatic beta-cells
Diabetes now affects about 20.8 million people in the United States. It is estimated that by the year 2050, approximately 48.3 million people in the United States will have diabetes [1]. Diabetes is generally classified into three separate categories; type 1 diabetes (also known as juvenile diabetes), type 2 diabetes (also known as adult onset diabetes), and gestational diabetes. The latter form occurs during pregnancy and is often reversible at the end of the pregnancy. Most of the diagnosed diabetes cases are of the type 2 form [2], and are now beginning to affect children as well as older adults. Though a complete understanding of the etiology of type 2 diabetes is lacking, it is often associated with phenomena such as insulin resistance and hyperglycemia. In the state of insulin resistance, beta-cells are still producing insulin. However, this insulin does not optimally perform its major function of facilitating glucose transport into such cells as muscle cells, fat cells (adipocytes), and endothelial cells.
Type 1 diabetes is similar to type 2 diabetes in that insulin is a major determinant in the disease. However, unlike type 2 diabetes, where insulin is produced until the latter stages of the disease, type 1 diabetes is typified by the lack of insulin production by the beta-cells. Therefore, people with type 1 diabetes, who represent about 10% of the diabetic population, must receive insulin in the form of injections or pumps.
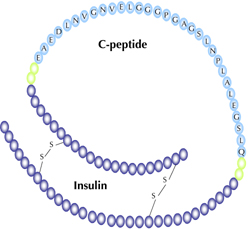
Insulin is produced in the islets of Langerhans as part of a molecule known as proinsulin, as shown in Figure 1. Two endopeptidases are involved in cleaving the proinsulin molecule, one cleaving exclusively on the C-terminal side of Arg 31/Arg 32 (B-chain/C-peptide junction), the other on the C-terminal side of Lys 64/Arg 65 of proinsulin (C-peptide/A-chain junction) [3]. Thus, this 31 amino acid peptide, when released, is in equimolar amounts to insulin [4]. Since the discovery of C-peptide [4], many groups have attempted to classify a biological role for this 31 amino acid peptide. For example, it has been established that C-peptide increases renal function [5-10], and microvascular blood flow in the skin of type 1 diabetic patients [11]. Other reports have demonstrated the ability of C-peptide to reduce diabetic complications associated with neuropathy [5, 12-14]. It has also been reported that human C-peptide [15], or its fragments [16], increases red blood cell (RBC) deformability in patients with type 1 diabetes.
 |
 |
Figure 1. Proinsulin C-peptide is shown with the amino acids representing C-peptide labeled. The four glutamic acid residues (E), and the aspartic acid residue (D), are thought to be key sites for C-peptide activity. |
|
Insulin-independent effects of C-peptide and metal activation
C-peptide has been shown by our group and other groups to interact with RBCs [15-18]. In the studies involving deformability [15-16], the RBC samples were incubated with C-peptide alone and, after an incubation period, improvements in deformability of the RBCs obtained from diabetic patients were measured.
In our lab, no measurable interactions between C-peptide and the RBC have been measured to date unless a metal (such as Zn2+) is added to the C-peptide prior to incubation with the RBC sample. At this point, we acknowledge that further studies are needed to explain the results obtained in our laboratory requiring metal activation. However, there are reports in the literature that contain data suggesting that C-peptide is more effective in the presence of higher concentrations of Zn2+. For example, in the aforementioned study involving C-peptide fragments and RBC deformability, addition of ethylenediaminetetraacetic acid (EDTA) to the samples prior to the addition of C-peptide abolished all deformability improvements [16]. Based on previous results involving a C-peptide-stimulated increase in the concentration of intracellular Ca2+, it was concluded that the EDTA was binding to Ca2+, and preventing the required increase in intracellular Ca2+ levels.
Another possibility is that the EDTA was competitively removing a metal from the C-peptide, thereby precluding its ability to interact with the RBC. In our studies, we have shown that C-peptide (when activated with Zn2+) results in an increase in ATP release when using a Ca2+-free buffer (submitted work). These results suggest that EDTA's ability to abolish activity may not have been solely due to a decrease in Ca2+, but also due to its ability to pull any activating metals from C-peptide. Previously, we have shown that other metals have the ability to stimulate interactions between C-peptide and the RBC [17]. Both Fe2+ and Cr3+ have been employed successfully in our studies.
We acknowledge that, to date, we are the only group requiring metal activation of C-peptide for any beneficial effects [17]. A simple answer would be that, in addition to the deformability studies involving C-peptide, we are one [17] of only a few groups [15, 16] to demonstrate beneficial effects with RBCs. Interestingly, even our first attempts at using C-peptide with RBCs were successful without the exogenous addition of a metal. However, through a careful mass spectroscopic analysis of our commercially purchased (>98% pure) C-peptide, it was discovered that there was a trace of Fe2+ bound to the C-peptide. When this sample was further purified in our own laboratories by high performance liquid chromatography (HPLC), reanalyzed and confirmed by mass spectrometry to not contain Fe2+, there were no subsequent effects from C-peptide on the RBCs. Furthermore, oxidation of that Fe2+ to Fe3+ resulted in minimal iron binding to the C-peptide, and subsequent loss of activity.
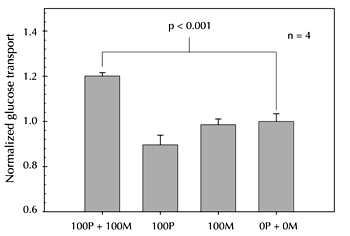
There are critiques of a hypothesis claiming that C-peptide requires metal activation for beneficial effects that are independent of insulin. The first and most obvious is the considerable amount of well-performed studies in the literature demonstrating C-peptide activity on multiple cell types without any exogenous addition of metal. Confirmation of C-peptide purity used by other groups in subsequent studies will provide clarity in this area. However, in addition to RBCs, our group can only elicit responses from other cell types when the C-peptide is in the presence of Zn2+. Figure 2 shows glucose uptake into endothelial cells obtained from bovine pulmonary artery in the presence of C-peptide, and C-peptide in the presence of Zn2+. As shown, the increased glucose uptake (measured as 14C-labelled glucose) is modest; nevertheless, in the absence of Zn2+, the measured increase is statistically equal to endothelial cells without any C-peptide added.
 |
|
Figure 2. Glucose uptake into cultured endothelial cells. Bovine pulmonary artery endothelial cells (bPAECs) were incubated with 1μCi/mL 14C-labeled glucose in a glucose-free physiological salt solution (PSS). After 60 min at 37°C/5%CO2, the bPAECs incubated in metal-activated C-peptide (P + M) showed approximately a 20% increase in radioactive counts per min (CPM) compared to those incubated with no metal-activated C-peptide. bPAECs incubated with only C-peptide (P) or Zn2+ (M), showed no significant difference in glucose transport. This data, normalized to the addition of buffer (0 nM C-peptide and 0 nM Zn2+), suggests that other cell types, in addition to RBCs, require metal activation for activity. All values are normalized to the CPM of the bPAECs incubated with no C-peptide or Zn2+. Error bars are ± SEM (n = 4). |
|
In addition to RBCs and endothelial cells, recent preliminary studies by our group clearly show that, in the presence of C-peptide, the ability of alveolar macrophages from rabbit lungs to phagocytose bacteria increases significantly due to an increase in glucose uptake through the macrophages GLUT1 transport mechanism.
Another critique is the notion that there are adequate levels of metals such as Fe2+ and Zn2+ in the bloodstream to activate C-peptide. While these metals do exist in the bloodstream, it is highly unlikely that they would exist in a bioavailable form. It is more likely that any Fe2+ or Zn2+ is bound to various other molecular species (e.g. hemes, proteins). Even if nanomolar levels of these metals do exist in a free form in the bloodstream, the millimolar levels of other metals (Na+, K+, etc.) would out-compete these less concentrated metals for binding sites (the glutamates and aspartate) on the C-peptide.
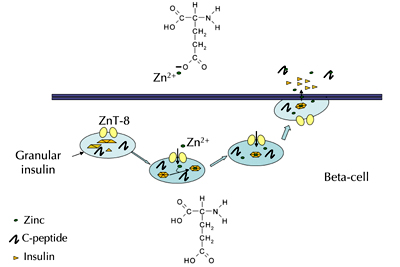
If C-peptide does require metal activation, it is not yet exactly clear how this activation takes place. However, an evaluation of pancreatic beta-cells and beta-cell granules holds a possible answer, which is summarized in Figure 3. The beta-cell granules contain Zn2+ transporter proteins that constantly shuttle Zn2+ into the granules. It is estimated that the concentrations of Zn2+ in these granules are well into the millimolar range. In the granules, Zn2+ is a key component of the insulin hexamer that exists in these granules. It has been reported that the Zn2+ facilitates insulin binding through histidine groups found on the insulin molecules, resulting in a hexameric complex that contains 6 insulin molecules and 2 Zn2+ atoms. Importantly, the estimated pH inside of the granules is approximately 5 [19].
 |
|
Figure 3. A possible mechanism for the origin of C-peptide interacting with Zn2+ upon secretion from the pancreatic beta-cell granules. In the granules (depicted as light blue circles), zinc (Zn2+) transporter proteins (ZnT-8) shuttle Zn2+ into the granules where it participates in the packaging of insulin as a crystalline hexamer. Also depicted in the granules is C-peptide; the pH inside the granules is estimated to be ca. 5. Upon secretion from the granules into the islets, the pH raises rapidly, resulting in the breakdown of the insulin hexamer into individual monomer units. The knowledge of C-peptide's role in this breakdown remains incomplete, although studies have shown that C-peptide does participate in the breakdown process. One possible mechanism is that the low pH of the granules keeps glutamic acid residues (structure shown inside the beta-cell) protonated until secretion where it changes to glutamate (structure shown above the beta-cell), a substance capable of binding Zn2+). |
|
In previous work, Kennedy et al. [20] show that a pH change (towards physiological pH) is a requirement for proper disaggregation of the insulin hexamer. In separate studies, Shafqat et al. [21] show that C-peptide has the ability to enhance the disaggregation of hexameric insulin into monomeric form. In the work by Shafqat, in vitro studies reveal an approximate 10-20% increase in monomeric insulin in the presence of C-peptide at pH 5; however, when insulin and C-peptide are added subcutaneously at the same depot to humans with type 1 diabetes, the increase in plasma insulin is approximately doubled between 10 and 60 minutes after injection in comparison to when the C-peptide and insulin were administered at different ports. Therefore, the studies by Kennedy and Shafqat suggest that, while there may be a reliance on pH for the breakdown of hexameric insulin, there appears to be a clear role for C-peptide in that process. One possibility is that the increase in pH deprotonates the glutamic acid and aspartic acid residues, rendering them ionized and more capable of binding Zn2+, and facilitating the breakdown of the insulin hexamer.
If C-peptide binding to Zn2+ was involved in the breakdown of the insulin hexamer, it would also be expected that insulin containing Zn2+ would be able to activate C-peptide-induced release of ATP from RBCs. In an in vitro study performed in our laboratory, we have been able to demonstrate that insulin containing 0.5% (w/w) Zn2+ added to C-peptide prior to incubation with RBCs did indeed elicit a response from these cells in the form of ATP release. However, similar studies, in which the insulin was purified by HPLC to contain no Zn2+ prior to adding to C-peptide, resulted in an ATP release value that was not statistically different from RBCs incubated in buffer or purified C-peptide alone (i.e., C-peptide with no exogenous metal added). These data suggest that C-peptide may be obtaining Zn2+ after secretion from the beta-cell granules.
Effects of C-peptide on cells in the bloodstream
The previous reports showing beneficial effects of C-peptide covering a wide range of complications (nephropathy, neuropathy, etc.) suggest the possible production of a multi-tasking molecule that is stimulated by the presence of C-peptide. It is well established that the endothelium-derived relaxing factor, nitric oxide (NO), behaves as such a multi-tasking molecule, being responsible for various physiological effects including smooth muscle relaxation, non-specific immune response, modulation in neurotransmission, and inhibition of platelet activation, aggregation and adhesion. NO produced by endothelial cells is also a major determinant in the control of vasodilation [22-24]. Physiologically, an alteration in shear stress to which the endothelial lining of blood vessels is subjected, has been suggested to be a major stimulus for NO release [23-26]. However, it has also been reported that, in the rabbit pulmonary circulation, in the absence of red blood cells (RBCs), alterations in shear stress alone did not evoke release of NO in the pulmonary circulation [27]. In contrast, in lungs perfused with rabbit blood, NO was found to be a determinant of vascular resistance. Importantly, the component of blood, responsible for the stimulation of endogenous NO synthesis, was determined to be the RBC, via the release of adenosine triphosphate (ATP) [28]. Indeed, the application of ATP to endothelial cells resulted in an increase in NO synthesis [29, 30]. ATP is of particular interest because it is present in millimolar amounts in RBCs [27, 28, 30, 31].
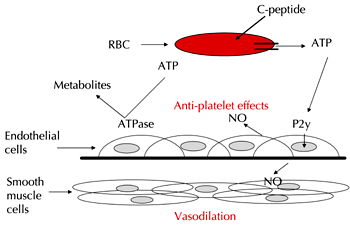
If C-peptide is a determinant of NO synthesis, and ATP is a required component for such synthesis, a hemodynamic response to C-peptide should be demonstrable in the RBC. Although previous studies have demonstrated that C-peptide had the ability to improve blood flow in skin and in the microvasculature, no mechanism for this improved blood flow had been reported. A recent publication reported that the ability of RBCs obtained from healthy rabbits to release ATP more than doubled over an 8 h incubation period, when incubated with C-peptide at physiological concentrations (1-10 nM) [17]. These results led us to believe that C-peptide may be exerting some of its effects via its ability to stimulate ATP release, which in turn promotes NO production, as shown in Figure 4.
 |
|
Figure 4. Proposed mechanism for C-peptide effects in the bloodstream. C-peptide results in the release of ATP from RBCs, which may participate in vessel dilation by stimulating NO production in the endothelium through purinergic receptor signaling. Furthermore, this ATP may stimulate NO production in platelets, inhibiting their ability to readily activate under normal conditions. |
|
In addition to the effects on RBCs obtained from non-diabetic animals (both rabbits and rats), C-peptide has also been shown to have a positive effect on RBCs obtained from rat models of both type 1 and type 2 diabetes, albeit with varying activities. A small scale study involving RBCs obtained from a rat model of type 1 diabetes revealed that the overall percentage increase in ATP release was not statistically different to the release measured from RBCs obtained from control rats [18]. In contrast, in a rat model of type 2 diabetes, ATP release from these cells were significantly less (approximately 50%) than that obtained from control RBCs [18]. Subsequent studies suggested that exogenously added C-peptide was interacting with the RBCs obtained from the type 2 rat models to a much lesser extent than the control RBCs (again, approximately a 50% reduction).
One of the longstanding criticisms of C-peptide is that people with type 2 diabetes, or at least those considered to be pre-diabetic or in the early stages of having been diagnosed with type 2 diabetes, often have normal or above-normal C-peptide levels in the bloodstream. Our studies, involving the RBCs obtained from type 1 (BB/Wor) rat models, suggests that these cells respond favorably to exogenously added C-peptide, and that there is no inherent problem with these cells that would preclude positive results from C-peptide replacement therapy. Administration of C-peptide to the type 2 RBC (especially when the RBC has been obtained from a poorly controlled type 2 subject, such as the BBZDR/Wor rat) results in less positive effects compared with type 1 or control RBCs. However, such results with type 2 RBCs should not lead to a premature conclusion that C-peptide does not have any beneficial physiological effects. On the contrary, it shows that C-peptide may be an important species in diabetes because, not unlike the interactions of insulin with certain cell types, C-peptide seems to find "resistance" from interacting with RBCs. Indeed, the concept of "resistance" against the C-peptide interaction with RBCs can also be supported by the excess of C-peptide often found in type 2 diabetics, which may actually bring about this resistance. Interestingly, it has been recently reported that metformin helps to overcome the reduced interactions between C-peptide and the type 2 RBC [18].
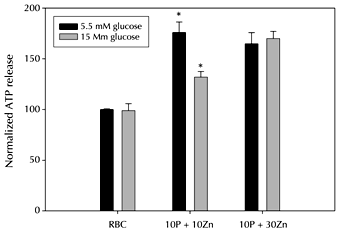
It should be noted that all of the studies involving Zn2+ activation of C-peptide have been performed in a somewhat conservative manner; that is, the Zn2+ has always been added in equimolar amounts to the C-peptide. Recently, we have hypothesized that the type 2 diabetic RBC (again, obtained from BBZDR/Wor-rats) exhibits some level of resistance to C-peptide due to an exposure of negatively charged phosphatidylserine (PS) lipids to the outer leaflet of the cell membrane. Such externalization of the PS lipid may repel the highly negatively charged C-peptide (with its five carboxylate groups, see Figures 1 and 2). If so, it would be likely that an increased ratio of Zn2+ to C-peptide would help overcome RBCs in hyperglycemic conditions more prone to PS lipid exposure. Figure 5, though preliminary, contains data supporting this notion. Figure 5 shows results from RBCs incubated in buffers containing incremental levels of glucose, and stimulated with C-peptide containing various levels of Zn2+. As shown, as the amount of Zn2+ added increases, the C-peptide is able to continue its role as a stimulus of ATP release from the RBC.
 |
|
Figure 5. ATP release from RBCs at different Zn2+:C-peptide ratios. The black bars represent ATP release in normal (5.5 mM) glucose buffer, while the white bars represent ATP release in high (15 mM) glucose buffer. The first set of bars show the ATP release from RBCs. The second set of bars show the ATP release in the presence of 10 nM C-peptide and 10 nM Zn2+. In normal glucose the ATP release increased 76% in the presence of Zn2+-activated C-peptide in comparison to RBCs alone. In high glucose, the metal-activated C-peptide resulted in an increase of only 32%. The third set of bars show the ATP release in the presence of 10 nM C-peptide and 30 nM Zn2+. There was no statistical difference in ATP release from the RBCs incubated in the normal and high glucose buffers. This data suggests that increasing the Zn2+:C-peptide ratio may help improve C-peptide interactions with the RBC, even under hyperglycemic conditions. (n = 4, C-pep = 10nM, Zn2+ = 10,30 nM, error = SEM, p < 0.002). |
|
C-peptide has been reported to stimulate improved blood flow in the forearm [32], skin [11], and myocardium [33] of patients with type 1 diabetes. Although C-peptide itself has been reported to stimulate eNOS in other cell types [32, 34], it may be possible that the improvements in the blood flow are due to the effects that C-peptide exerts on RBCs.
As mentioned above, it is now well-established that RBC-derived ATP is a recognized stimulus of NO production in the endothelium due to its binding of P2Y purinergic receptors found on the endothelium. We have now shown that C-peptide is able to stimulate ATP release from the RBC. Collectively, these findings suggest that C-peptide's ability to improve overall blood flow may be due to its ability to increase the release of NO-stimulating ATP.
The effects of C-peptide in this regard may be multifaceted, as well. That is, there are many stimuli for increasing ATP release from the RBC including hypoxia [35-38], pharmaceutical agents (e.g., iloprost) [39], acidosis [31], and deformation [28, 40-42]. We have learned that C-peptide (when metal-activated) is able to increase the release of ATP from RBCs even in the absence of any flow-induced deformation. Although a mechanism for this release has not been completely elucidated, an increase of glucose uptake into the RBC must occur before the ATP release is measured. This hypothesis is in agreement with a recent finding suggesting that increased glycolytic metabolism by the RBC facilitates an increase of ATP release from these cells [43]. Therefore, in addition to stimulating ATP release in the absence of any external stimuli, the ability of C-peptide to improve the deformability of the RBC may coincide with a mechanism proposed by Sprague [40, 44-46]. This mechanism describes a deformation-induced activation of a G-protein coupled receptor that subsequently leads to the release of ATP release from the RBC.
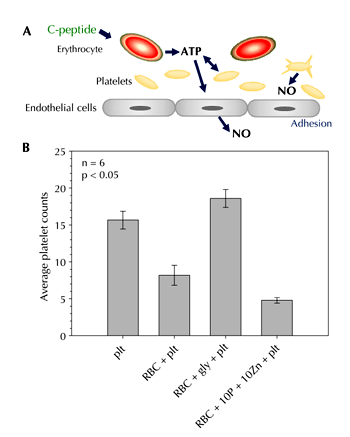
Recently, in vitro studies involving immobilized endothelial cells revealed that C-peptide may also have an effect on platelet adhesion [47]. This finding is in contrast to a previous report that C-peptide does not have any direct effects on platelets [48]. In this context, we have also found no direct effects of C-peptide on platelets. However, when platelets are added to a sample of RBCs containing C-peptide, the platelet behavior is altered. Specifically, in a recent study, platelets were pumped across endothelial cells immobilized in flow channels having a diameter of about 200 micrometers [47]. The number of platelets adhering to this endothelium per unit area depended upon the amount of ATP present in the media with the platelets. As shown in Figure 6, when these platelets were added to a solution of RBCs, and then pumped across the endothelium, the number of platelets adhering decreased from 16 ± 2 to 8 ± 3. Inhibition of ATP release from these RBCs resulted in the number of platelets adhering to increase to a value of 18 ± 2. These studies suggested that the RBC-derived ATP release was able to affect platelet adherence to the endothelium by stimulating NO production in both the endothelium and the platelets. As a powerful inhibitor of platelet activation, NO production may have reduced platelet adhesion to an endothelium. Indeed, when C-peptide was added to the RBCs prior to addition of the platelets, the number of platelets adhering to the endothelium decreased to a value of 5 ± 1. Consistent with previous work, C-peptide in the absence of Zn2+ had no effect on platelet adhesion.
 |
|
Figure 6. Determination of platelet adhesion to an immobilized endothelium under various conditions. The upper illustration (A) depicts a proposed series of events that may lead to platelet adhesion, and the ability of C-peptide to alter that adhesion through the stimulated release of ATP from RBCs. In the diagram (B), the first bar represents platelets (plt) alone adhering to an endothelium under conditions of flow. The second bar is another aliquot of those platelets flowing across the endothelium, but in the presence of RBCs; note the decrease in platelets adhering to the endothelium. In the third bar, the ATP release from the RBCs was inhibited with glybenclamide. This led to an increase in platelet adhesion number. The final bar depicts the platelets flowing over the endothelium in the presence of RBCs that had been incubated with Zn2+-activated C-peptide. C-peptide alone or Zn2+ alone had no effect on platelet adhesion. These data suggest that C-peptide-induced ATP release from RBCs may have a beneficial effect on the reduction of platelet adhesion in vivo. |
|
In addition to the adhesion studies performed above using the RBCs from healthy rabbits, our group has also performed experiments involving platelets and RBCs obtained from rat models of type 1 diabetes. These studies (currently in revision) demonstrated that NO production is increased in platelets obtained from type 1 rats when stimulated with the supernatant from centrifuged RBCs from the same animals that had been stimulated with metal-activated C-peptide. Further studies confirmed that RBC metal activation was required, and that ATP was the source of stimulus in the supernatant. Previously, it had been shown that administration of C-peptide caused a significant delay in arteriolar and venular thrombus growth in normal and diabetic mice [49]. However, subsequent perfusion with insulin (100 microU/ml) reversed these effects, leading us to believe that C-peptide may not be beneficial as an anti-thrombotic treatment option. In contrast to these studies demonstrating a beneficial effect of C-peptide on platelet activation, studies by Hu et al. [48] (in which insulin or C-peptide, or a combination thereof, was administered to whole blood) show that insulin results in an increase in fibrinogen binding; however, C-peptide demonstrates no ability to reverse this trend [50].
In addition to platelets, C-peptide has also been reported to decrease leukocyte adhesion [50]. When C-peptide was administered to rats previously administered L-NAME to inhibit NO production, a significant increase in leukocyte rolling and adhered leukocytes were measured. However, in the presence of C-peptide alone, these measured leukocyte effects were attenuated. It was concluded that these effects were related to the ability of C-peptide to affect endothelial cell adhesion molecules, and maintenance of NO release from the endothelium.
C-peptide: a multi-tasking molecule or stimulus of such?
To date, C-peptide has been reported to induce a number of various activities in various cell types, and a multitude of beneficial in vivo effects, suggesting that C-peptide is truly a multi-tasking molecule in its own right. However, the question remains as to whether C-peptide is directly responsible for these effects or if it is stimulating the production of another molecule, such as ATP, that is stimulating these effects.
Previous studies involving C-peptide interactions with RBCs clearly demonstrate that C-peptide is able to stimulate glucose uptake into the RBC, resulting in the release of ATP into the extracellular matrix [17, 18]. This glucose uptake results in an increase of ATP production by the RBC. An increase in cellular energy (ATP), due to this glucose uptake, would result in an increase in kinase activity, due to ATP serving as the phosphate source for the phosphorylation activity of kinases. In addition, it would also serve as the energy for ATPase driven pumps.
In addition to its effects on the intracellular machinery, we strongly believe that one of C-peptide’s major roles is its role in ATP release from RBCs. As mentioned above, the studies involving C-peptide and its effect on RBC deformability inspired much of our current efforts involving C-peptide. There are many diseases, in addition to diabetes, where the RBC-derived ATP release is smaller than in healthy individuals. All of these diseases have associated blood flow problems. People with cystic fibrosis [40], primary pulmonary hypertension [51], and diabetes [18, 52, 53] all have RBCs that release less ATP than those RBCs obtained from healthy controls. It is also established that people with diabetes have RBCs that are less deformable than those of healthy patients [54, 55]. If C-peptide has the ability to improve this reduced deformability, it is likely that it will increase RBC-derived ATP, as these cells flow through resistance vessels in the intact circulation [40].
Recent data has also suggested that ATP may be a determinant in platelet activity in vivo [47]. ATP acts on platelets through the P2X receptor, which regulates a calcium ion channel in platelets. The P2X receptor, and hence ATP, has been reported to not participate in platelet activation or subsequent aggregation steps. However, recently, our group has demonstrated that ATP does indeed participate in platelet activity. In fact, it was shown that below normal levels of ATP binding to the P2X receptor resulted in a decrease in NO production by platelets, resulting in an increase in platelet aggregation [47]. However, as ATP levels increased, both the NO production and aggregation rates of platelets decreased. As the ATP levels continued increasing, the platelets again began to aggregate. This trend suggested that below normal and above normal levels of ATP resulted in an increase in platelet aggregation. The key feature of these studies, in relation to diabetic complications, is that people with type 1 diabetes, having minimal to no C-peptide in the bloodstream, would likely have below normal levels of RBC-derived ATP available to stimulate platelet NO. This results in platelets with above-normal activity.
Concluding thought and perspective direction of future work
One of the main questions still requiring further studies is whether or not C-peptide requires metal activation. The answer to this question will become more clear with time. However, even if C-peptide does require activation, it still needs to be elucidated if the key feature for cellular response is the metal or C-peptide itself. For example, it has been reported that both the full length C-peptide and its fragments demonstrate activity on RBCs. Our group has been able to reproduce similar studies involving the full length peptide and its fragments (submitted work). Specifically, we have shown that both the full length peptide and its fragments stimulate ATP release from RBCs; however, even the fragments will only elicit this response when bound to zinc. Other studies by our group have shown that the metal alone elicits no response from the RBCs, even at low micromolar levels [17], and this trend also holds for other cell types, as shown in Figure 2.
It is quite clear that C-peptide exerts effects on cells. It seems that almost all of these effects are beneficial. While previous studies have demonstrated that C-peptide affects intracellular mechanisms, its ability to stimulate ATP release from RBCs enables C-peptide to affect intercellular communication [56, 57], and mechanisms. To date, our group has demonstrated that RBC-derived ATP, stimulated by C-peptide, can increase NO production in endothelial cells and platelets, and affect platelet adhesion to endothelial cells.
In our opinion, C-peptide may be to the components of the bloodstream as insulin is to other tissues. Studies to date performed in our laboratory seem to suggest that C-peptide strongly affects cells whose main glucose transporter is GLUT1. Importantly, insulin is not a determinant in glucose uptake through GLUT1. However, future work will be required to determine the mechanism by which the glucose increase into cells is occurring. In summary, future work should focus on elucidating whether diabetes is indeed a single (insulin) or multiple (insulin and C-peptide) disease. If activation of C-peptide with Zn2+ is validated by other groups, it will be more facile to determine the roles of each substance in diabetic complications.
Disclosures: The authors report no conflict of interests.
Acknowledgments:
This work was funded by the National Institutes of Health (DK 071888).
References
- Venkat NK, Boyle JP, Geiss LS, Saaddine JB, Thompson TJ. Impact of recent increase in incidence on future diabetes burden. Diabetes Care 2006. 29(9):2114-2115. [DOD] [CrossRef]
- Amos AF, McCarty DJ, Zimmet P. The rising global burden of diabetes and its complications: estimates and projections by 2010. Diabet Med 1997. 14:S5-S85. [DOD]
- Davidson HW, Rhodes CJ, Hutton JC. Intraorganellar calcium and pH control proinsulin cleavage in the pancreatic cell via two distinct site-specific endopeptidases. Nature 1988. 333:93-96. [DOD] [CrossRef]
- Rubenstein AH, Clark JL, Melani F, Steiner DF. Secretion of proinsulin c-peptide by pancreatic b cells and its circulation in blood. Nature 1969. 224(5220):697-699. [DOD] [CrossRef]
- Johansson BL, Borg K, Fernqvist-Forbes E, Kernell A, Odergren T, Wahren J. Beneficial effects of C-peptide on incipient nephropathy and neuropathy in patients with type 1 diabetes mellitus. Diabet Med 2000. 17(3):181-189. [DOD] [CrossRef]
- Johansson BL, Kernell A, Sjoberg S, Wahren J. Influence of combined C-peptide and insulin administration on renal function and metabolic control in diabetes type 1. J Clin Endocr Metab 1993. 77(4):976-981. [DOD] [CrossRef]
- Johansson BL, Sjoeberg S, Wahren J. The influence of human C-peptide on renal function and glucose utilization in type 1 (insulin-dependent) diabetic patients. Diabetologia 1992. 35(2):121-128. [DOD] [CrossRef]
- Rebsomen L, Pitel S, Boubred F, Buffat C, Feuerstein JM, Raccah D, Vague P, Tsimaratos M. C-peptide replacement improves weight gain and renal function in diabetic rats. Diabetes Metab 2006. 32(3):223-228. [DOD] [CrossRef]
- Samnegard B, Jacobson SH, Jaremko G, Johansson BL, Sjoquist M. Effects of C-peptide on glomerular and renal size and renal function in diabetic rats. Kidney Int 2001. 60(4):1258-1265. [DOD] [CrossRef]
- Sjoquist M, Huang W, Johansson BL. Effects of C-peptide on renal function at the early stage of experimental diabetes. Kidney Int 1998. 54(3):758-764. [DOD] [CrossRef]
- Forst T, Kunt T, Pohlmann T, Goitom K, Engelbach M, Beyer J, Pfutzner A. Biological activity of C-peptide on the skin microcirculation in patients with insulin-dependent diabetes mellitus. J Clin Invest 1998. 101(10):2036-2041. [DOD] [CrossRef]
- Stevens M, Zhang W, Li F, Sima AA. C-peptide corrects endoneurial blood flow but not oxidative stress in type 1 BB/Wor rats. Am J Physiol Endocrinol Metab 2004. 287(3):E497:E505. [DOD]
- Ido Y, Vindigni A, Chang K, Stramm L, Chance R, Heath WF, DiMarchi RD, Di Cera E, Williamson JR. Prevention of vascular and neural dysfunction in diabetic rats by C-peptide. Science 1997. 277(5325):563-566. [DOD] [CrossRef]
- Zhang W, Kamiya H, Ekberg K, Wahren J, Sima AA. C-peptide improves neuorpathy in type 1 diabetic BB/Wor rats. Diabetes Metab Res 2007. 23:63-70. [DOD] [CrossRef]
- Kunt T, Schneider S, Pfutzner A, Goitum K, Engelbach M, Schauf B, Beyer J, Forst T. The effect of human proinsulin C-peptide on erythrocyte deformability in patients with type I diabetes mellitus. Diabetologia 1999. 42(4):465-471. [DOD] [CrossRef]
- Hach T, Forst T, Kunt T, Ekberg K, Pfutzner A, Wahren J. C-peptide and its C-Terminal fragments improve erythrocyte deformability in type 1 diabetes patients. Exp Diabetes Res 2008. 2008:730594. [DOD]
- Meyer JA, Froelic JM, Reid GE, Karunarathne W, Spence DM. Metal-activated C-peptide facilitates glucose clearance and the release of a nitric oxide stimulus via the GLUT1 transporter. Diabetologia 2008. 51(1):175-182. [DOD] [CrossRef]
- Meyer JA, Subasinghe W, Sima AA, Keltner Z, Reid GE, Daleke D, Spence DM. Zinc-activated C-peptide resistance to the type 2 diabetic erythrocyte is associated with hyperglycemia-induced phosphatidylserine externalization and reversed by metformin. Mol Biosyst 2009. 5:1157-1162. [DOD] [CrossRef]
- Hutton JC. The internal pH and membrane potential of the insulin-secretory granule. Biochem J 1982. 204(1):171-178. [DOD]
- Kennedy RT, Huang L, Aspinwall CA. Extracellular pH is required for rapid release of insulin from Zn-insulin precipitates in beta-cell secretory vesicles during exocytosis. J Am Chem Soc 1996. 118(7):1795-1796. [DOD] [CrossRef]
- Shafqat J, Melles E, Sigmundsson K, Johansson BL, Ekberg K, Alvelius G, Henriksson M, Johansson J, Wahren J, Joernvall H. Proinsulin C-peptide elicits disaggregation of insulin resulting in enhanced physiological insulin effects. Cell Mol Life Sci 2006. 63:1805-1811. [DOD] [CrossRef]
- Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980. 288:373-376. [DOD] [CrossRef]
- Ignarro LJ, Buga G, Dhaudhuri G. EDRF generation and release from perfused bovine pulmonary artery and vein. Eur J Pharmacol 1988. 149:79-88. [DOD] [CrossRef]
- Palmer R, Ferrige MJ, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxation factor. Nature 1987. 327:524-526. [DOD] [CrossRef]
- Buga GM, Gold ME, Fukuto JM, Ignarro LJ. Shear stress-induced release of nitric oxide from endothelial cells grown on beads. Hypertension 1991. 17:187-193. [DOD]
- Rubanyi GM, Romero JC, Vanhoutte PM. Flow-induced release of endothelium-derived relaxing factor. Am J Physiol 1986. 250:H1145-H1149. [DOD]
- Sprague RS, Stephenson AH, Dimmit RA, Weintraub NA, Branch CA, McMurdo L, Lonigro AJ. Effect of L-NAME on pressure-flow relationships in isolated rabbit lungs: Role of red blood cells. Am J Physiol 1995. 296:H1941-H1948. [DOD]
- Sprague RS, Ellsworth ML, Stephenson AH, Lonigro AJ. ATP: the red blood cell link to NO and local control of the pulmonary circulation. Am J Physiol 1996. 271:H2717-H2722. [DOD]
- Dull RO, Tarbell JM, Daves PF. Mechanism of flow-mediated signal transduction in endothelial cells: kinetics of ATP surface concentrations. J Vasc Res 1992. 29:410-419. [DOD] [CrossRef]
- Bogle RG, Coade SB, Moncada S, Pearson JD, Mann GE. Bradykinin and ATP stimulate L-arginine uptake and nitric oxide release in vascular endothelial cells. Biochem Biophys Res Commun 1991. 80:926-932. [DOD] [CrossRef]
- Bergfeld GR, Forrester T. Release of ATP from human erythrocytes in response to a brief period of hypoxia and hypercapnea. Cardiovasc Res 1992. 26:40-47. [DOD] [CrossRef]
- Johansson BL, Wahren J, Pernow J. C-peptide increases forearm blood flow in patients with type 1 diabetes via a nitric oxide-dependent mechanism. Am J Physiol Endocrinol Metab 2003. 285:E864-E870. [DOD]
- Hansen A, Johansson BL, Wahren J, Von Bibra H. C-peptide exerts beneficial effects on myocardial blood flow and function in patients with type 1 diabetes. Diabetes 2002. 51(10):3077-3082. [DOD] [CrossRef]
- Li H, Xu L, Dunbar JC, Dhabuwala CB, Sima AA. Effects of C-peptide on expression of eNOS and iNOS in human cavernosal smooth muscle cells. Urology 2004. 64(3):622-627. [DOD] [CrossRef]
- Collins DM, McCullough WT, Ellsworth ML. Conducted vascular responses: communication across the capillary bed. Microvasc Res 1998. 56:43-53. [DOD] [CrossRef]
- Ellsworth ML, Forrester T, Ellis CG, Dietrich HH. The erythrocyte as a regulator of vascular tone. Am J Physiol 1995. 269:H2155-H2161. [DOD]
- McCullough WT, Collins DM, Ellsworth ML. Arteriolar responses to extracellular ATP in striated muscle. Am J Physiol 1997. 272:H1886-H1891. [DOD]
- Faris AN, Spence DM. Measuring the simultaneous effects of hypoxia and deformation on ATP release from erythrocytes. Analyst 2008. 133(5):678-684. [DOD] [CrossRef]
- Genes LI, Tolan NV, Hulvey MK, Martin RS, Spence DM. Adressing a vascular endothelium array with blood components using underlying microfluidic channels. Lab Chip 2007. 7(10):1256-1259. [DOD] [CrossRef]
- Sprague RS, Ellsworth ML, Stephenson AH, Kleinhenz ME, Lonigro AJ. Deformation-induced ATP release from red blood cells requires cystic fibrosis transmembrane conductance regulator activity. Am J Physiol 1998. 275:H1726-H1732. [DOD]
- Sprague RS, Ellsworth ML, Stephenson AH, Lonigro AJ. Increases in flow rate stimulate adenosine triphosphate release from red blood cells in isolated rabbit lungs. Exp Clin Cardiol 1998. 3:73-77. [DOD]
- Fischer DJ, Torrence NJ, Sprung RJ, Spence DM. Determination of erythrocyte deformability and its correlation to cellular ATP release using microbore tubing with diameters that approximate resistance vessels in vivo. Analyst 2003. 128:1163-1168. [DOD] [CrossRef]
- Cao Z, Bell JB, Mohanty JG, Nagababu E, Rifkind JM. Nitrite enhances RBC hypoxic ATP synthesis and the release of ATP into the vasculature: a new mechanism for nitrite-induced vasodilation. Am J Physiol Heart Circ Physiol 2009. 297:H1494-H1503. [DOD] [CrossRef]
- Olearczyk JJ, Stephenson AH, Lonigro AJ, Sprague RS. Receptor-mediated activation of the heterotrimeric G-protein Gs results in ATP release from erythrocytes. Med Sci Monit 2001. 7(4):669-674. [DOD]
- Olearczyk JJ, Stephenson AH, Lonigro AJ, Sprague RS. Heterotrimeric G protein Gi is involved in a signal transduction pathway for ATP release from erythrocytes. Am J Physiol 2004. 286(3 Pt 2):H940-H945. [DOD]
- Sprague RS, Ellsworth ML, Stephenson AH, Lonigro AJ. Participation of cAMP in a signal-transuction pathway relating erythrocyte deformation to ATP release. Am J Physiol Cell Physiol 2001. 281(4):C1158-C1164. [DOD]
- Karunarathne W, Ku CJ, Spence DM. The dual nature of extracellular ATP as a concentration-dependent platelet P2X1 agonist and antagonist. Integr Biol (Camb) 2009. 1(11-12):655-663. [DOD]
- Hu H, Li N, Ekberg K, Johansson BL, Hemjdahl P. Insulin, but not proinsulin C-peptide, enhances platelet fibrinogen binding in vitro in Type 1 diabetes mellitus patients and healthy subjects. Thromb Res 2002. 106(2):91-95. [DOD] [CrossRef]
- Lindenblatt N, Braun B, Menger MD, Klar E, Vollmar B. C-peptide exerts antithrombotic effects that are repressed by insulin in normal and diabetic mice. Diabetologia 2006. 49(4):792-800. [DOD] [CrossRef]
- Scalia R, Coyle KM, Levine BJ, Booth G, Lefer AM. C-peptide inhibits leukocyte-endothelium interaction in the microcirculation during acute endothelial dysfunction. FASEB J 2000. 14:2357-2364. [DOD] [CrossRef]
- Sprague RS, Stephenson AH, Ellsworth ML, Keller C, Lonigro AJ. Impaired release of ATP from red blood cells of humans with primary pulmonary hypertension. Exp Biol Med 2001. 226(5):434-439. [DOD]
- Carroll JS, Subasinghe W, Raththagala M, Baguzis S, Oblak T, Root PD, Spence D. An altered erythrocyte pentose phosphate pathway affects the ability of red cells to release ATP, a nitric oxide stimulus. Mol Biosyst 2006. 2:305-311. [DOD] [CrossRef]
- Sprague RS, Stephenson AH, Bowles EA, Stumpf MS, Lonigro AJ. Reduced expression of Gi in erythrocytes of humans with type 2 diabetes is associated with impairment of both cAMP generation and ATP release. Diabetes 2006. 55(12):3588-3593. [DOD] [CrossRef]
- Srour MA, Bilto YY, Juma M. Susceptibility of erythrocytes from non-insulin-dependent diabetes mellitus and hemodialysis patients, cigarette smokers and normal subjects to in vitro oxidative stress and loss of deformability. Clin Hemorheol Microcirc 2000. 22(3):173-180. [DOD]
- Ghigo D, Bosia A, Pagani A, Pescarmona GP, Pagano G, Lenti G. Changes in erythrocyte deformability resulting from alterations of redox metabolism in diabetes mellitus. Res Clin Lab 1983. 13(Suppl 3):375-381. [DOD]
- Carroll J, Ku CJ, Karunarathne W, Spence D. Red blood cell stimulation of platelet nitric oxide production indicated by quantitative monitoring of the communication between cells in the bloodstream. Anal Chem 2007. 79:5133-5138. [DOD] [CrossRef]
- Ku CJ, D'Amico Oblak T, Spence DM. Interactions between multiple cell types in parallel microfluidic channels: monitoring platelet adhesion to an endothelium in the presence of an anti-adhesion drug. Anal Chem 2008. 80(19):7543-7548. [DOD] [CrossRef]
This article has been cited by other articles:
|


)
)
)
)
)
)



