Review
| Rev Diabet Stud,
2009,
6(3):180-186 |
DOI 10.1900/RDS.2009.6.180 |
C-Peptide in the Vessel Wall
Daniel Walcher1, Nikolaus Marx2
1Department of Internal Medicine II, Cardiology, University of Ulm, Germany
2Department of Internal Medicine I, Cardiology, University of Aachen, Germany
Address correspondence to: Nikolaus Marx, e-mail: nmarx@ukaachen.de
Manuscript submitted October 9, 2009; resubmitted October 17, 2009; accepted October 27, 2009.
Keywords: diabetes, C-peptide, vascular cell, arteriosclerosis, inflammation, reactive oxygen species, smooth muscle cell, lipoprotein
Abstract
Patients with insulin resistance and early type 2 diabetes exhibit an increased sensitivity to develop a diffuse and extensive pattern of arteriosclerosis leading to a remarkable increase in vascular complications, including myocardial infarction and stroke. The accelerated atherosclerosis in these patients is likely to be multifactorial. In this review, we introduce the new hypothesis that C-peptide could play a role as a mediator of lesion development. Patients with type 2 diabetes show increased levels of the proinsulin cleavage product C-peptide, and in the past few years, various groups have examined the effect of C-peptide in vascular cells as well as its potential role in lesion development. Recent data suggest that C-peptide deposits in the vessel wall could promote the recruitment of monocytes and CD4-positive lymphocytes in early arteriosclerotic lesions. Furthermore, C-peptide induces proliferation of vascular smooth muscle cells, a critical step in atherogenesis and restenosis formation. The present review summarizes this new pathophysiological aspect and discusses the potential relevance for lesion development.
Introduction
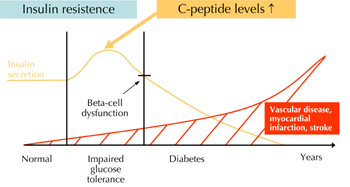
Patients with diabetes and insulin resistance exhibit an increased sensitivity to develop arteriosclerosis with its sequelae acute myocardial infarction and stroke [1]. These patients temporarily demonstrate elevated levels of C-peptide, the cleavage product of proinsulin (Figure 1). For a long time, C-peptide has been considered to be a biologically inert substance without any physiological function. Recent work in kidney cells suggests that C-peptide can activate intracellular signaling pathways such as Na+-K+-ATPase [2]. In addition, C-peptide activates various signaling pathways in different cell-types [3, 4] suggesting that C-peptide may be biologically active.
 |
 |
Figure 1. Development of arteriosclerosis in diabetic patients. Patients with diabetes exhibit an increased risk to develop vascular diseases with their sequelae myocardial infarction and stroke. During the "prediabetic" state of insulin resistance these patients temporarily demonstrate elevated levels of insulin and C-peptide. Recent experimental data suggest that C-peptide may play a causal role in early atherogenesis in these patients. |
|
Some groups have shown that C-peptide administration in type 1 diabetes results in amelioration of diabetes-induced renal and nerve dysfunction. A C-peptide treatment for 6 months improved sensory nerve function [5, 6]. Furthermore, beneficial effects by C-peptide replacement on renal function and structure in type 1 diabetes were reported [7, 8]. Given that C-peptide increases capillary blood flow in type 1 diabetic patients, Wallerath et al. demonstrated in vitro that C-peptide stimulates the release of NO from endothelial NO synthase in endothelial cells, and that this effect is mediated by induction of Ca2+ influx into the cells [9]. In the kidney, C-peptide supplementation suppresses diabetes-induced abnormal renal eNOS expression. This downregulation can be an explanation for C-peptide's beneficial effects on diabetic nephropathy [10].
The present review focuses on C-peptides effects in vascular cells and discusses the potential relevance of these effects on atherogenesis in patients with type 2 diabetes.
Atherogenesis
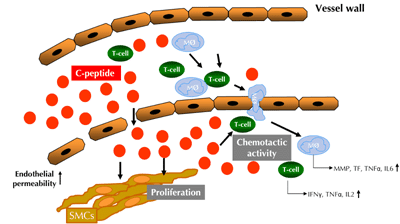
Atherogenesis is currently understood as an inflammatory process in the vessel wall with different phases and stages [11]. The early phase of atherogenesis is characterized by endothelial dysfunction caused by various risk factors like smoking, diabetes, hypertension or hypercholesterolemia. In addition, the endothelium is activated in this phase, releasing chemotactic proteins such as MCP-1, a monocyte chemoattractant, or RANTES and IP-10, both chemoattractants for lymphocytes. These chemokines attract inflammatory cells, mainly monocytes and CD4-positive lymphocytes, to adhere to endothelial adhesion molecules, with subsequent migration of these cells into the vessel wall. Monocytes, once migrated into the subendothelial space, become macrophages, expressing scavenger receptors on their surface. These macrophages mediate the uptake of lipoprotein particles such as oxidized low-density lipoprotein (LDL). Through lipid uptake, these cells differentiate to foam cells, the classic cellular substrate of arteriosclerotic lesions. The macrophage foam cells then express additional proinflammatory and proatherogenic mediators such as cytokines, matrix metalloproteinases, and reactive oxygen species. They also secrete procoagulant molecules such as tissue factor (Figure 2). All these factors act on other cells in the vessel wall and thus promote lesion development [12, 13].
 |
|
Figure 2. Potential role of C-peptide in early atherogenesis in patients with insulin resistance and early type 2 diabetes mellitus. C-peptide deposits in the intima and through its chemotactic activity on monocytes and CD4+ lymphocytes during endothelial dysfunction with increased endothelial permeability. This facilitates the recruitment of inflammatory cells into the vessel wall and perpetuate the proinflammatory process. Furthermore, C-peptide can activate smooth muscle cell proliferation. T-cell: CD4-positive lymphocytes. Mø: monocytes/macrophages. SMC: smooth muscle cell. MMP: matrix metalloproteinase. TF: tissue factor. |
|
The second inflammatory cell type recruited during endothelial dysfunction is the CD4-positive lymphocyte. These cells enter the vessel wall as naïve Th0 cells [14]. In the subendothelial space, these cells then encounter antigens like oxidized LDL and differentiate towards Th1 cells, which release pro-inflammatory cytokines such as IFNγ, TNFα and IL-2. Some of these cytokines then enhance endothelial expression and release of T cell-specific chemokines, creating a vicious cycle of cell activation and cell recruitment [15]. In addition, these Th1 cytokines activate other cells in the vessel wall, namely macrophages and smooth muscle cells (SMCs), which orchestrates the inflammatory response in the vessel wall. With increased recruitment of these inflammatory cells, fatty streaks develop and SMCs from the media start to proliferate and migrate into the intima. As lesion formation progresses, advanced and potentially complicated lesions are formed. These lesions may form plaque and lead to a progressive narrowing of the vessel lumen, which potentially creates persistent angina if located in the coronary artery. Alternatively, the plaques may become vulnerable and upon rupture can cause an acute coronary artery syndrome [16].
C-peptide is present in the vascular wall of atherosclerotic lesions
Patients with metabolic syndrome or type 2 diabetes mellitus exhibit an increased propensity for the development of a scattered and extensive pattern of arteriosclerosis [1]. Typically, these insulin resistant patients demonstrate increased serum levels of insulin, and C-peptide which is a cleavage product of proinsulin. C-peptide is released into the blood stream in amounts equimolar to insulin.
Over the past few decades, the role of insulin in atherogenesis has been examined by various groups. It has been suggested that insulin may promote lesion development, for example, by inducing proliferation of vascular SMCs [17]. Others demonstrated that insulin exhibits anti-inflammatory and antiatherogenic properties in vascular cells such as monocytes [18, 19]. Overall, the role of insulin in atherogenesis remains unclear. Presently and most important, we lack clinical and epidemiologic evidence to show that insulin may have harmful effects with respect to macrovascular events.
For a long time, C-peptide has been considered to be biologically inert, but recent data suggest that C-peptide binds to specific yet unidentified, cell surface receptors. Recently, our group hypothesized that C-peptide might deposit in the vessel wall in these patients in early atherogenesis [20]. Immunohistochemical analyses of early arteriosclerotic lesions of patients with diabetes from the PDAY Study (Patho-biological Determinants of Atherosclerosis in Youth) revealed that C-peptide deposition takes place mainly in the subendothelial space and the intima. Some of the examined diabetic patients also exhibited C-peptide deposition in the media. In contrast, only very little C-peptide deposition has been found in early arteriosclerotic lesions of non-diabetic subjects. Computer-assisted analyses revealed significantly higher C-peptide deposition in lesions from diabetic individuals compared with lesions of non-diabetic subjects matched by age, sex and risk-factors. Interestingly, in both diabetic and non-diabetic subjects, deposition of insulin or pro-insulin was not detectable.
C-peptide colocalizes with inflammatory cells in the vessel wall
Staining of parallel sections as well as immunofluorescence techniques demonstrated colocalization of C-peptide with intimal monocyte/macrophages and CD4-positive lymphocytes in some of the diabetic individuals [20, 21]. In the studies cited above, C-peptide deposition has been found in 100% of the 21 diabetic individuals examined, while monocyte infiltration was only present in 77%, and CD4-positive lymphocyte infiltration only in 57%. These data suggested that C-peptide deposition may precede monocyte and T cell migration into the vessel wall. Based on this observation, the hypothesis was raised that C-peptide may deposit in the vessel wall during early atherogenesis and then—through chemotactic effects—promote the recruitment of monocytes and CD4-positive lymphocytes.
C-peptide - a peptide with chemotactic activity?
In vitro migration assays, employing a modified Boyden chamber, revealed that C-peptide induces the migration of both monocytes and CD4+ lymphocytes. Interestingly, insulin did not have such an effect. Stimulation of monocytes with C-peptide-induced cell migration in a concentration-dependent manner with a maximal augmentation of 2.3 ± 0.4 at 1 nmol/l. The extent of C-peptide-induced monocyte migration resembled the effect of the established monocyte chemokine MCP-1, which increased monocyte migration by 2.9 ± 0.2 fold. Stimulation of CD4+ cells with C-peptide induced cell migration in a concentration-dependent manner with a maximal induction of 2.1 ± 0.5 at 10 nmol/l. The extent of C-peptide-induced CD4+ cell migration was similar to the effect of an established T cell chemokine (RANTES), which led to a 2.1 ± 0.6 fold increase in cell migration. It is interesting that the combined stimulation of CD4+ cells with C-peptide and RANTES did not have an additive migratory effect. In addition, checkerboard analyses showed that C-peptide induces chemotaxis rather than chemokinesis [20, 21].
Also, C-peptide did not induce the migration of human neutrophils—cells not present in arteriosclerotic lesions. This underscored the specificity of C-peptide's chemotactic effects on monocytes and lymphocytes. However, no migratory effect of C-peptide was seen on B cells, another subset of lymphocytes [22].
C-peptide activates intracellular signaling pathways in inflammatory cells
Inhibition migration assays in monocytes and CD4-positive lymphocytes, western blot analyses, and activity assays, all demonstrated that C-peptide mediates its chemotactic activity through an unidentified pertussis toxin-sensitive G-protein coupled receptor, with subsequent downstream activation of PI3-kinase γ.
Previous work from other groups has shown that Src-kinase is involved in interferon-gamma-inducible protein-10 (IP-10) mediated chemotactic response of lymphocytes [23]. In our experiments, PP2 (a specific inhibitor of Src-kinase) and transfection of Src siRNA, abolished C-peptide-induced T-cell migration, suggesting that C-peptide also signals through this pathway. Additional experiments demonstrated that PI3-kinase activation led to the involvement of small Rho-GTPases, like RhoA, Rac-1, and Cdc42 in these cells. Furthermore, C-peptide stimulated phosphorylation of p21 activated kinase (PAK), LIM domain-containing protein kinase (LIMK), and cofilin downstream of Rac-1 and Cdc42, leading to cofilin inactivation and actin filament stabilization. On the other hand, C-peptide activates Rho kinase (ROCK) and myosin light chain (MLC) phosphorylation downstream of RhoA, thereby stimulating myosin-mediated cell contraction [22]. These data support an active role of C-peptide in chemotaxis of inflammatory cells.
In summary, these data suggest that C-peptide may deposit in the arterial intima in patients with insulin resistance and early type 2 diabetes during endothelial dysfunction with increased endothelial permeability. Subsequently, this results in the attraction of monocytes and CD4-positive lymphocytes into the vessel wall. Such mechanisms may promote lesion development and potentially explain why patients with diabetes develop such a diffuse and extensive pattern of arteriosclerosis at a very early point in time.
C-peptide promotes the differentiation of monocytes/macrophages
In addition to the chemotactic effect, interesting data exists that in monocyte-like ThP1 cells C-peptide increases the expression of the PPARγ-regulated gene CD36, an important scavenger receptor for the macrophage uptake of oxidized LDL in arteriosclerotic lesions [24]. These data suggest that C-peptide may also promote the differentiation of monocyte/macrophages towards foam cells, thus representing another potential pro-atherogenic effect of C-peptide.
C-peptide is internalized in vascular cells
Recent reports showed that C-peptide is internalized in the cytoplasm of HEK-293 and Swiss 3T3 cells. In addition, Lindahl et al. demonstrated C-peptide transport into nuclei using a labeled peptide [25]. The internalization was followed at 37°C for up to 1 h, and was reduced at 4°C after preincubation with pertussis toxin. It is concluded that this occurs via an energy-dependent pertussis toxin-sensitive mechanism. Recently, C-peptide internalization was found in vascular cells, like endothelial cells and vascular smooth muscle cells. C-peptide was detected as punctuate intracellular structures and these structures were identified as early endosomes. From these results the authors concluded that endosomes could be a signaling station, through which C-peptide might achieve its cellular effects [26]. Similar results we found in monocytes and CD4-positive cells (Walcher et al., unpublished data).
C-peptide's proatherogenic role in smooth muscle cell proliferation
Vascular smooth muscle cells play a critical role in the development of arteriosclerotic plaques by proliferating and subsequently moving from the media into early lesions and fatty streaks [11]. VSMCs are also important in restenosis formation after coronary intervention. After vascular injury, these cells start to proliferate and then they migrate into the developing neointima. Thus, they become the major cellular substrate of the restenotic tissue [27]. Strategies to modulate VSMC proliferation after intervention, e.g. by the implantation of drug-eluting stents, reduce neointima formation and restenosis [28]. This underscores the pathophysiological relevance of VSMC proliferation in these processes. Several mechanisms, like platelet-derived growth factor (PDGF) release from activated platelets, secretion of cytokines, and growth factors from inflammatory cells, have all been shown to induce VSMC proliferation during atherogenesis and restenosis formation [11]. Since C-peptide also co-localizes with SMCs in the media of early arteriosclerotic lesions in some diabetic subjects, it has been suggested that C-peptide could also exhibit biological activity in these cells [15].
Conflicting data exist on the role of C-peptide in SMC proliferation. A recent report by Kobayashi et al. demonstrated that 3 days of treatment with human C-peptide under high glucose concentrations resulted in the inhibition of rat SMC proliferation [29]. These effects observed at high C-peptide concentrations (100 nmol/l) were mediated by an inhibition of PDGF-beta receptor expression and a reduction in p42/p44 MAP-kinase phosphorylation. In contrast, our own group demonstrated an induction of SMC proliferation upon C-peptide stimulation [30]. In our experimental setting, human C-peptide induced the proliferation of human aortic SMCs, while rat C-peptide exhibited similar effects in rat aortic SMCs.
Moreover, C-peptide stimulation of VSMCs activates Src- and PI-3 kinase and leads to downstream phosphorylation of the MAPK ERK1/2. Previous work has shown that Src-kinase is involved in LDL-induced VSMC proliferation [31]. Recently, activation of PI-3 kinase by C-peptide has been demonstrated in various cell types including vascular cells like monocytes and CD4-positive lymphocytes, suggesting that this signaling molecule plays a critical role in C-peptide-induced cell activation in different cell types. The downstream control of VSMC proliferation by extracellular stimuli takes place in the mid- to late G1 phase of the cell cycle, where D-type cyclins promote G1- to S-phase transition, which leads to Rb phosphorylation [32, 33]. Our data, showing an increase in cyclin D1 expression as well as Rb phosphorylation suggest that C-peptide acts via similar signaling pathways [30].
In addition to its chemotactic effect on monocytes and CD4-positive lymphocytes, C-peptide could enhance VSMC proliferation in the vasculature. It would thus promote both the development of arteriosclerotic lesions and neointima formation after coronary intervention. Future studies in animal models will be needed to evaluate the pathophysiological relevance of C-peptide-induced VSMC proliferation in atherogenesis and neointima formation in vivo.
C-peptide: conflicting data in type 1 and type 2 diabetes?
The majority of data described above suggest that C-peptide may promote lesion development in patients with type 2 diabetes mellitus and insulin resistance, while the application of C-peptide in type 1 diabetic patients who lack C-peptide has been shown to improve diabetic microvascular complications such as diabetic neuropathy. The potential proatherogenic action of C-peptide is not in conflict with the clinical benefits of C-peptide treatment in patients with type 1 diabetes. From other clinical disorders, like hypo- and hyperthyroidism, we know that treatment with L-thyroxine in patients with hypothyroidism is beneficial, but elevated levels of L-thyroxine in those with hyperthyroidism can cause serious clinical manifestations. Comparing situations, supplementation of C-peptide in type 1 diabetic patients may be beneficial, whereas in patients with insulin resistance and type 2 diabetes, increased levels of C-peptide may be harmful.
Further studies in animal models of arteriosclerosis are warranted to examine whether the hypothesis of C-peptide's proatherogenic effects holds true in vivo. Moreover, additional work is needed to identify the C-peptide receptor, in order to modulate the physiological function of C-peptide.
Conclusions and perspectives
C-peptide might contribute to the initiation and progression of lesion development in diabetic subjects. Further understanding of these mechanisms may pave the way for future therapies to target vascular disease in patients with diabetes and as such reduce cardiovascular morbidity and mortality in this high risk population.
Disclosures: The authors report no conflict of interests.
Acknowledgments:
This work was supported by grants from the Deutsche Forschungsgemeinschaft (SFB 451, project B9 and B11, MA2047/4-1, WA2145/1-1) to Prof. Dr. Nikolaus Marx and to Dr. Daniel Walcher, as well as a grant from the Else-Kröner Fresenius Stiftung to Dr. Daniel Walcher.
References
- Beckman JA, Creager MA, Libby P. Diabetes and atherosclerosis: epidemiology, pathophysiology, and management. JAMA 2002. 287(19):2570-2581. [DOD] [CrossRef]
- Wahren J, Ekberg K, Johansson J, Henriksson M, Pramanik A, Johansson BL, Rigler R, Jörnvall H. Role of C-peptide in human physiology. Am J Physiol Endocrinol Metab 2000. 278(5):E759-E768. [DOD]
- Kitamura T, Kimura K, Jung BD, Makondo K, Okamoto S, Canas X, Sakane N, Yoshida T, Saito M. Proinsulin C-peptide rapidly stimulates mitogen-activated protein kinases in Swiss 3T3 fibroblasts: requirement of protein kinase C, phosphoinositide 3-kinase and pertussis toxin-sensitive G-protein. Biochem J 2001. 355(Pt 1):123-129. [DOD] [CrossRef]
- Kitamura T, Kimura K, Jung BD, Makondo K, Sakane N, Yoshida T, Saito M. Proinsulin C-peptide activates cAMP response element-binding proteins through the p38 mitogen-activated protein kinase pathway in mouse lung capillary endothelial cells. Biochem J 2002. 366(Pt 3):737-744. [DOD]
- Ekberg K, Brismar T, Johansson BL, Jonsson B, Lindström P, Wahren J. Amelioration of sensory nerve dysfunction by C-peptide in patients with type 1 diabetes. Diabetes 2003. 52(2):536-541. [DOD] [CrossRef]
- Ekberg K, Brismar T, Johansson BL, Lindström P, Juntti-Berggren L, Norrby A, Berne C, Arnqvist HJ, Bolinder J, Wahren J. C-peptide replacement therapy and sensory nerve function in type 1 diabetic neuropathy. Diabetes Care 2007. 30(1):71-76. [DOD] [CrossRef]
- Samnegard B, Jacobson SH, Jaremko G, Johansson BL, Ekberg K, Isaksson B, Eriksson L, Wahren J, Sjöquist M. C-peptide prevents glomerular hypertrophy and mesangial matrix expansion in diabetic rats. Nephrol Dial Transplant 2005. 20(3):532-538. [DOD] [CrossRef]
- Sjöquist M, Huang W, Johansson BL. Effects of C-peptide on renal function at the early stage of experimental diabetes. Kidney Int 1998. 54(3):758-764. [DOD] [CrossRef]
- Wallerath T, Kunt T, Forst T, Closs EI, Lehmann R, Flohr T, Gabriel M, Schäfer D, Göpfert A, Pfützner A, Beyer J, Förstermann U. Stimulation of endothelial nitric oxide synthase by proinsulin C-peptide. Nitric Oxide 2003. 9(2):95-102. [DOD] [CrossRef]
- Kamikawa A, Ishii T, Shimada K, Makondo K, Inanami O, Sakane N, Yoshida T, Saito M, Kimura K. Proinsulin C-peptide abrogates type-1 diabetes-induced increase of renal endothelial nitric oxide synthase in rats. Diabetes Metab Res Rev 2008. 24(4):331-338. [DOD] [CrossRef]
- Ross R. Atherosclerosis - an inflammatory disease. N Engl J Med 1999. 340(2):115-126. [DOD] [CrossRef]
- Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med 2005. 352(16):1685-1695. [DOD] [CrossRef]
- Libby P, Geng YJ, Aikawa M, Schoenbeck U, Mach F, Clinton SK, Sukhova GK, Lee RT. Macrophages and atherosclerotic plaque stability. Curr Opin Lipidol 1996. 7(5):330-335. [DOD] [CrossRef]
- Robertson AK, Hansson GK. T cells in atherogenesis: for better or for worse? Arterioscler Thromb Vasc Biol 2006. 26(11):2421-2432. [DOD] [CrossRef]
- Marx N. Diabetes und Arteriosklerose. Diabetes und Stoffwechsel 2003. 4:205-211. [DOD]
- Libby P, Simon DI. Inflammation and thrombosis: the clot thickens. Circulation 2001. 103(13):1718-1720. [DOD]
- Trovati M, Anfossi G. Influence of insulin and of insulin resistance on platelet and vascular smooth muscle cell function. J Diabetes Complications 2002. 16:35-40. [DOD] [CrossRef]
- Dandona P, Chaudhuri A, Ghanim H, Mohanty P. Anti-inflammatory effects of insulin and pro-inflammatory effects of glucose: relevance to the management of acute myocardial infarction and other acute coronary syndromes. Rev Cardiovasc Med 2006. 7:S25-S34. [DOD] [CrossRef]
- Dandona P, Chaudhuri A, Mohanty P, Ghanim H. Anti-inflammatory effects of insulin. Curr Opin Clin Nutr Metab Care 2007. 10:511-517. [DOD] [CrossRef]
- Marx N, Walcher D, Raichle C, Aleksic M, Bach H, Grüb M, Hombach V, Libby P, Zieske A, Homma S, Strong J. C-peptide colocalizes with macrophages in early arteriosclerotic lesions of diabetic subjects and induces monocyte chemotaxis in vitro. Arterioscler Thromb Vasc Biol 2004. 24(3):540-545. [DOD] [CrossRef]
- Walcher D, Aleksic M, Jerg V, Hombach V, Zieske A, Homma S, Strong J, Marx N. C-peptide induces chemotaxis of human CD4-positive cells: involvement of pertussis toxin-sensitive G-proteins and phosphoinositide 3-kinase. Diabetes 2004. 53(7):1664-1670. [DOD] [CrossRef]
- Aleksic M, Walcher D, Giehl K, Bach H, Grüb M, Durst R, Hombach V, Marx N. Signalling processes involved in C-peptide-induced chemotaxis of CD4-positive lymphocytes. Cell Mol Life Sci 2009. 66(11-12):1974-1984. [DOD] [CrossRef]
- Kukhtina NB, Arefieva TI, Krasnikova TL. Intracellular signal cascade in CD4+ T-lymphocyte migration stimulated by interferon-gamma-inducible protein-10. Biochemistry (Mosc) 2005. 70:652-656. [DOD] [CrossRef]
- Al-Rasheed NM, Chana RS, Baines RJ, Willars GB, Brunskill NJ. Ligand-independent activation of peroxisome proliferator-activated receptor-gamma by insulin and C-peptide in kidney proximal tubular cells: dependent on phosphatidylinositol 3-kinase activity. J Biol Chem 2004. 279(48):49747-49754. [DOD] [CrossRef]
- Lindahl E, Nyman U, Melles E, Sigmundsson K, Stahlberg M, Wahren J, Obrink B, Shafqat J, Joseph B, Jörnvall H. Cellular internalization of proinsulin C-peptide. Cell Mol Life Sci 2007. 64(4):479-486. [DOD] [CrossRef]
- Luppi P, Geng X, Cifarelli V, Drain P, Trucco M. C-peptide is internalised in human endothelial and vascular smooth muscle cells via early endosomes. Diabetologia 2009. 52(10):2218-2228. [DOD] [CrossRef]
- Bittl JA. Advances in coronary angioplasty. N Engl J Med 1996. 335(17):1290-1302. [DOD] [CrossRef]
- Stone GW, Ellis SG, Cox DA, Hermiller J, O'Shaughnessy C, Mann JT, Turco M, Caputo R, Bergin P, Greenberg J, Popma JJ, Russell ME. One-year clinical results with the slow-release, polymer-based, paclitaxel-eluting TAXUS stent: the TAXUS-IV trial. Circulation 2004. 109(16):1942-1947. [DOD] [CrossRef]
- Kobayashi Y, Naruse K, Hamada Y, Nakashima E, Kato K, Akiyama N, Kamiya H, Watarai A, Nakae M, Oiso Y, Nakamura J. Human proinsulin C-peptide prevents proliferation of rat aortic smooth muscle cells cultured in high-glucose conditions. Diabetologia 2005. 48:2396-2401. [DOD] [CrossRef]
- Walcher D, Babiak C, Poletek P, Rosenkranz S, Bach H, Betz S, Durst R, Grüb M, Hombach V, Strong J, Marx N. C-Peptide induces vascular smooth muscle cell proliferation: involvement of SRC-kinase, phosphatidylinositol 3-kinase, and extracellular signal-regulated kinase 1/2. Circ Res 2006. 99(11):1181-1187. [DOD] [CrossRef]
- Cho HM, Choi SH, Hwang KC, Oh SY, Kim HG, Yoon DH, Choi MA, Lim S, Song H, Jang Y, Kim TW. The Src/PLC/PKC/MEK/ERK signaling pathway is involved in aortic smooth muscle cell proliferation induced by glycated LDL. Mol Cells 2005. 19(1):60-66. [DOD]
- Beijersbergen RL, Bernards R. Cell cycle regulation by the retinoblastoma family of growth inhibitory proteins. Biochim Biophys Acta 1996. 1287(2-3):103-120. [DOD]
- Harbour JW, Dean DC. Rb function in cell-cycle regulation and apoptosis. Nat Cell Biol 2000. 2(4):E65-E67. [DOD] [CrossRef]
This article has been cited by other articles:
|


)
)


