Chapter II. Diabetic Nephropathy
| Rev Diabet Stud,
2015,
12(1-2):134-156 |
DOI 10.1900/RDS.2015.12.134 |
Targeting Mitochondria and Reactive Oxygen Species-Driven Pathogenesis in Diabetic Nephropathy
Runa Lindblom1,2,3,4, Gavin Higgins1,2,45,, Melinda Coughlan1,2,3,6,7, Judy B. de Haan2,78,
1Glycation, Nutrition and Metabolism Laboratory, Baker IDI Heart and Diabetes Institute, Melbourne, Victoria, Australia
2Diabetic Complications Division, Baker IDI Heart and Diabetes Institute, Melbourne, Victoria, Australia
3Department of Medicine, Central Clinical School, Monash University, Melbourne, Victoria, Australia
4Equal contribution by first authors
5Department of Biochemistry and Molecular Biology, Monash University, Clayton, Victoria, Australia
6Department of Epidemiology and Preventive Medicine, Monash University, Melbourne, Victoria, Australia
7Equal contribution by senior authors
8Oxidative Stress Laboratory, Baker IDI Heart and Diabetes Institute, Melbourne, Victoria, Australia
Address correspondence to: Judy B. de Haan, Head, Oxidative Stress Laboratory, Diabetic Complications Division, Baker IDI Heart and Diabetes Institute, 75 Commercial Road, Melbourne, Victoria 3004, Australia, e-mail: judy.dehaan@bakeridi.edu.au
Manuscript submitted March 30, 2015; accepted April 15, 2015.
Keywords: diabetic nephropathy, reactive oxygen species, apoptosis, necrosis, mitochondrial dysfunction
Abstract
Diabetic kidney disease is one of the major microvascular complications of both type 1 and type 2 diabetes mellitus. Approximately 30% of patients with diabetes experience renal complications. Current clinical therapies can only mitigate the symptoms and delay the progression to end-stage renal disease, but not prevent or reverse it. Oxidative stress is an important player in the pathogenesis of diabetic nephropathy. The activity of reactive oxygen and nitrogen species (ROS/NS), which are by-products of the diabetic milieu, has been found to correlate with pathological changes observed in the diabetic kidney. However, many clinical studies have failed to establish that antioxidant therapy is renoprotective. The discovery that increased ROS/NS activity is linked to mitochondrial dysfunction, endoplasmic reticulum stress, inflammation, cellular senescence, and cell death calls for a refined approach to antioxidant therapy. It is becoming clear that mitochondria play a key role in the generation of ROS/NS and their consequences on the cellular pathways involved in apoptotic cell death in the diabetic kidney. Oxidative stress has also been associated with necrosis via induction of mitochondrial permeability transition. This review highlights the importance of mitochondria in regulating redox balance, modulating cellular responses to oxidative stress, and influencing cell death pathways in diabetic kidney disease. ROS/NS-mediated cellular dysfunction corresponds with progressive disease in the diabetic kidney, and consequently represents an important clinical target. Based on this consideration, this review also examines current therapeutic interventions to prevent ROS/NS-derived injury in the diabetic kidney. These interventions, mainly aimed at reducing or preventing mitochondrial-generated oxidative stress, improving mitochondrial antioxidant defense, and maintaining mitochondrial integrity, may deliver alternative approaches to halt or prevent diabetic kidney disease.
Abbreviations: 8-OHdG – 8-hydroxy-2'-deoxyguanosine; 8-oxodG – 8-oxo-7,8-dihydro-2'-deoxyguanosine; ADP – adenosine diphosphate; AGE – advanced glycation end-product; AIF – apoptosis-inducing factor; aPC – activated protein C; ATM – ataxia telangiectasia mutated; ATP – adenosine triphosphate; ATR – ataxia telangiectasia and Rad3-related; BER – base excision repair; BMF – Bcl-2 modifying factor; CKD – chronic kidney disease; CVD – cardiovascular disease; DIABLO – direct IAP-binding protein with low pI; EMT – endothelial-mesenchymal transition; EndoG – endonuclease G; ER – endoplasmic reticulum; ERK – extracellular signal-regulated kinase; ESRD – end-stage renal disease; ETC – electron transport chain; FADD – FAS-associated protein with death domain; GPx1 – glutathione peroxidase 1; H2O2 – hydrogen peroxide; ICR – Institute of Cancer Research; IDH2 – isocitrate dehydrogenase 2; IMS – intermembrane space; MAPK – mitogen-activated protein kinase; MCP-1 – monocyte chemotactic protein 1; MIF – macrophage migration inhibitory factor; MIOX – myo-inositol oxygenase; MOMP – mitochondrial outer membrane permeability; mPTP – mitochondrial permeability transition pore; mtDNA – mitochondrial DNA; MTH1 – mut T homolog 1; MUTYH – mut Y homolog; NADPH – nicotinamide adenine dinucleotide phosphate hydrogen; NF-κB – nuclear factor kappa-light-chain-enhancer of activated B cells; •NO2- – nitrogen dioxide; NOX – NADPH-oxidase; NRK – normal rat kidney; O2•- – superoxide; OGG1 – oxoguanine glycosylase; OH• – hydroxyl radical; ONOO- – peroxynitrite; OPG – osteoprotegerin; OXPHOS – oxidative phosphorylation; PARP – poly (ADP-ribose) polymerase; PKC – protein kinase C; RAGE – receptor for AGEs; RAS – renin-angiotensin system; ROS/NS - reactive oxygen and nitrogen species; Smac – second mitochondrial-derived activator of caspases; Smad2 – mothers against decapentaplegic homolog 2; SOD2 – superoxide dismutase 2; STZ – streptozotocin; TCA – tricarboxylic acid; TGF – transforming growth factor; TRAIL – tumor necrosis factor-related apoptosis-inducing ligand; VCAM-1 – vascular cell adhesion molecule 1; ZDF – Zucker diabetic fatty
1. Introduction
Diabetes is recognized as the leading cause of end-stage renal disease (ESRD) in developed countries [1]. Statistics show a steady increase in the global prevalence of type 2 diabetes, with the worldwide obesity epidemic recognized as a major risk factor [2]. With an estimated 7.7% of the world population aged between 20-79 predicted to be diabetic by 2030, microvascular complications such as diabetic nephropathy are expected to contribute to a significant percentage of the worldwide morbidity rate [3]. Furthermore, chronic kidney disease (CKD), which affects approximately 5-30% of people with type 2 diabetes, is a significant risk factor for the development of cardiovascular disease (CVD) [4, 5]. Indeed, diabetic patients with microalbuminuria are 2-4 times more likely to experience cardiovascular impairments than non-diabetic patients [6], whilst those diabetic patients with overt kidney disease show a 4-8-fold increased risk of cardiovascular disease [7]. Despite the administration of glucose-, lipid-, and blood pressure-lowering drugs, nearly half of all diabetic patients continue on to develop renal and cardiovascular complications. Thus, the search for effective therapies is still a major goal in the treatment of diabetic patients.
Oxidative stress and inflammation, induced by chronic elevations in blood glucose in diabetic patients, are increasingly being recognized as risk factors [8] and significant mechanistic contributors [9-11] inseparably linked with the development of diabetic complications including CKD and ultimately ESRD. There is evidence that reactive oxygen and nitrogen species (ROS/NS) play key roles in the pathogenesis of diabetic nephropathy. This has initiated increased efforts to reduce or prevent the cumulative damage of ROS/NS-induced injury. However, clinical antioxidant trials with primary endpoints of CVD reduction have shown little success, which emphasizes the need for more knowledge about the major sites of ROS/NS production and the processes they are involved in [12-14].
Increased attention is currently paid to the significance of mitochondrial ROS/NS generated in response to elevated glucose level as the major player in the process of cell death. This impacts not only the ability of a cell to function optimally, but ultimately and collectively it dictates how an organ like the diabetic kidney responds to its challenging high-glucose environment. This review will focus on the latest evidence, both clinically and pre-clinically, to evaluate the contribution of ROS/NS to acute kidney failure in diabetes. Therapies that specifically target mitochondrial ROS to prevent cell death will be discussed and newer intervention options around this paradigm will be presented.
2. Contribution of ROS/NS production to diabetic nephropathy
Oxidative stress by definition is due to an overproduction of ROS/NS and/or a deficiency in enzymatic and non-enzymatic antioxidant defense, such that the balance is tipped in favor of ROS/NS accumulation and cell damage. Evidence from preclinical studies supports the role of ROS/NS involvement in the pathogenesis of diabetic CKD and ESRD. It has been shown that elevated levels of markers for ROS/NS damage accumulate in diabetic kidneys [15-18] and, under high-glucose conditions mimicking diabetes, in various kidney-derived cultured cells [19, 20]. Data from our laboratory have clearly demonstrated increased oxidative stress and advanced kidney disease in type 1 diabetic mice lacking the major cytosolic and mitochondrial antioxidant enzyme glutathione peroxidase 1 (GPx1) [21]. In particular, our study demonstrated a strong link between increased oxidative stress, enhanced inflammatory response (monocyte chemotactic protein 1 (MCP-1), vascular cell adhesion molecule 1 (VCAM-1)), and increased pro-fibrotic mediators (transforming growth factor (TGF) β, collagens I and III) in the diabetic kidney cortex. Indeed, the ROS/NS, peroxynitrite, has been shown to play a key role in the pathogenesis of the diabetic glomerular lesion [22]. Other preclinical studies with genetic alterations of the major ROS/NS producing enzymes, the NADPH-oxidases (NOX), in particular NOX4, lend further support to the theory of oxidant-mediated diabetic kidney injury [23]. Ablation of NOX4 in a mouse model of type 1 diabetes caused marked protection from both structural and functional kidney damage [9]. Thus, targeting NOX4 with pharmacological inhibitors may offer a viable therapeutic strategy to reduce diabetic kidney injury.
Clinically, type 2 diabetes patients demonstrate elevations of excreted urinary 8-hydroxy-2'-deoxyguanosine (8-OHdG), a marker of oxidative DNA damage, which was positively correlated with the extent of tubulointerstitial kidney injury in these patients [24]. Furthermore, a complementary marker of DNA damage, 8-oxo-7,8-dihydro-2'-deoxyguanosine (8-oxodG), strongly predicted the progression of the disease in type 2 diabetics over a 5-year follow-up period [25].
At the molecular level, ROS/NS, produced as a consequence of elevated glucose levels, affect cell signaling pathways associated with metabolism, cell proliferation, and cell death [26]. Of particular relevance to diabetic kidney disease, a number of studies have shown the involvement of ROS in glucose-mediated activation of the protein kinase C (PKC) pathway in mesangial cells, leading to increased TGF-β levels [27, 28], an important mediator of kidney fibrosis [29, 30]. ROS have also been shown to activate NF-κB signaling in mesangial cells, causing a pro-inflammatory response [31].
Evidence from antioxidant studies has further supported a role for ROS/NS-mediated modulations of cellular signaling. Indeed, our own studies with the antioxidant and GPx1 mimetic ebselen have shown reductions in the activation of p38 MAP kinases (MAPK) and JNK in normal rat kidney (NRK) cells [21], suggesting the involvement of ROS/NS in these pathways linked to diabetic kidney disease. Others have used disparate antioxidants, and found a reduction in TGF-β-stimulated ROS and endothelial-mesenchymal transition (EMT), as well as lower levels of phosphorylated Smad2, p38 MAPK, and extracellular signal-regulated kinase (ERK) in renal tubular epithelial cells [32]. In conclusion, there is sufficient preclinical evidence supporting the role of glucose-stimulated ROS/NS in disrupting the relevant signal transduction cascades and transcription factors involved in cell function and metabolism, leading to glomerular mesangial expansion and tubulointerstitial fibrosis.
3. Mitochondrial ROS/NS in the pathogenesis of diabetic nephropathy
The importance of intact, efficiently functioning mitochondria is imperative to the prevention of kidney damage and renal failure [33, 34]. Mitochondria are involved in many biological processes, such as energy production, calcium homeostasis, and the regulation of cell death pathways, including apoptosis, programmed cell death, and unregulated necrosis [35]. Mitochondria play paradoxical roles in fueling cellular homeostasis through ATP produced by oxidative phosphorylation (OXPHOS), and in initiating cell death via the release of intermembrane space cell death proteins, such as cytochrome c, Smac/DIABLO, apoptosis-inducing factor (AIF), and endonuclease G (EndoG). These actions can be linked to an imbalance in redox regulation within the mitochondrial matrix, an association that plays a significant role in diabetic nephropathy. This line of evidence will be further discussed in the next sections.
OXPHOS involves the transfer of electrons across complexes I-IV of the electron transport chain (ETC) to generate a membrane potential (Δψm) that is essential to establish the proton motive force driving ATP production. While OXPHOS is critical to meet the energy requirements of all eukaryotic cells, this is most evident in proximal and distal tubule cells that depend on ATP to drive Na+/K+ antiporters and Ca2+ transporters, respectively [36-38]. However, the byproduct of OXPHOS is the production of superoxide (O2•-) that occurs as a result of electrons escaping from complex I and III and reacting with molecular oxygen. In intact mitochondria, complex I and the Q1 site of complex III release O2•- toward the mitochondrial matrix, whilst the Q0 site of complex III releases O2•- toward the intermembrane space. [39, 40]. Impairment of complex I can result in an increase in the formation of reactive species and the development of renal damage similar to that seen in diabetic nephropathy, as demonstrated in Ndufs6 knockout mice [41]. Moreover, O2•- can be converted into other ROS/NS, such as hydrogen peroxide (H2O2) and hydroxyl radicals (OH•), and can react with nitric oxide to form peroxynitrite (ONOO-) and nitrogen dioxide (•NO2-) [33]. For proximal tubules, this is thought to be exacerbated under hyperglycemic conditions, where an increase in glucose reabsorption may result in increased glycolysis and OXPHOS, resulting in increased oxidative stress [34]. In addition, methylglyoxal, an -dicarbonyl byproduct of glycolysis, can induce the formation of advanced glycation end-products (AGEs) that exacerbate ROS production [42].
AGEs accumulate during diabetes. They are associated with an increase in oxidative stress through signaling via the receptor for AGEs (RAGE) [33, 43]. Our group has demonstrated that RAGE-induced cytosolic ROS/NS promotes mitochondrial O2•- production via activation of complex I of the mitochondrial ETC, triggering mitochondrial permeability transition pore (mPTP) formation and cell death, which can be attenuated in diabetic RAGE-/- mice [44]. Formation of AGEs through methylglyoxal accumulation results in modification of subunits, core protein 1 and cytochrome c1, of complex III causing O2•- production, as shown in the cortex of diabetic rat kidneys [45]. Overexpression of glyoxalase I, the enzyme responsible for detoxifying methylglyoxal, has been shown to reduce the expression of complex I, II, and III, while reducing ROS production and apoptosis under hyperglycemic conditions in mouse mesangial cells [46]. In conclusion, existing evidence argues in favor of a significant role for ROS/NS produced as byproducts of the ETC in diabetic nephropathy. Indeed, Brownlee and coworkers proposed the unified hypothesis of diabetic complications where ETC-generated ROS/NS induce diabetic complications [47-49]. Current thinking has expanded on these ideas to speculate that the actions of ETC-generated ROS/NS most likely involve additional pathways and ROS/NS-producing enzymes. These aspects are further discussed below.
The regulation of redox homeostasis within the diabetic milieu has been the subject of two recent studies on diabetic nephropathy. The tubular-specific enzyme myo-inositol oxygenase (MIOX) was identified as a regulator of redox imbalance in the mitochondria under hyperglycemic conditions [50]. These studies showed an increase in MIOX in the renal cortex of streptozotocin (STZ)-induced diabetic ICR (Institute of Cancer Research) outbred male mice. Moreover, an increase in ROS and the incidence of apoptosis associated with increased MIOX expression was observed in the kidney proximal tubule cell line, LLC-PK1 [50]. Likewise, the redox enzyme p66Shc, which localizes within mitochondria, has been implicated in the regulation of ROS/NS production and apoptosis during exposure to increased levels of glucose in diabetic nephropathy [51]. This enzyme has been shown to be epigenetically suppressed by the coagulation protease, activated protein C (aPC), in podocytes in diabetic nephropathy [51].
A significant focus of recent research has been on the NADPH oxidases within the renal cortex in the diabetic milieu, particularly NOX4, as discussed above. Block et al. demonstrated that NOX4 is upregulated under hyperglycemic conditions within the mitochondrial compartment of the cell [52]. NOX4 has been localized within renal cortex membrane fractions [52]. Since NOX isoenzymes reside within cells and span membranes of cells [53], it is likely that NOX4 spans the inner membrane of mitochondria, and that O2•- or H2O2 are released into the intermembrane space (IMS) of mitochondria. Initially, it had been proposed that NOX4 produces H2O2 only [54], but it is also capable of producing O2•-, as shown by Block et al. [52].
NOX4 has also been localized in the mitochondrial fraction of kidney glomerular mesangial cells that are known to be primary targets for glucose-mediated oxidative injury [23]. While NADP+/NADPH can traverse the outer mitochondrial membrane, it is impermeable to the inner membrane. NOX4 requires NADPH to become active. The fact that functional O2•--producing NOX4 has been identified in diabetic mesangial cells suggests that the carboxyl-teminus of NOX4 is positioned in contact with the intermembrane space [52]. Furthermore, NOX4 may indirectly influence the citric acid cycle, which is located within the mitochondrial matrix, through its influence on isocitrate metabolism. This includes the conversion of isocitrate into α-ketoglutarate by isocitrate dehydrogenase 2 (IDH2), which is dependent on NADP+ [55]. Recently, the use of metabolomics has revealed that NOX4 plays a key role in regulating the tricarboxylic acid (TCA) urinary metabolite fumarate in diabetic Akita mice by exerting its effect on the mitochondrial enzyme fumarate hydratase. Importantly, interventions with a NOX1/NOX4 inhibitor restored fumarate hydratase and fumarate levels and reduced albuminuria, glomerular hypertrophy, and mesangial matrix accumulation in these diabetic mice [56]. Nonetheless, it is evident that O2•- produced by mitochondrial NOX4 cannot have a direct link to deficiencies in the ETC or mitochondrial DNA (mtDNA) integrity that are housed within the inner mitochondrial membrane or matrix respectively, unless converted to membrane permeable molecules such as H2O2 or ONOO-, which we have demonstrated to be increased in the diabetic kidney [57].
If NOX4 has any deleterious influence within the mitochondria then it is likely to occur within the IMS. It is also likely that glutathione reductase competes with NOX4 for NADPH to maintain the balance of glutathione used for Gpx1 and Gpx4 within the mitochondria to detoxify H2O2 by conversion into H2O. Indeed, the NOX inhibitor apocynin has been shown to restore the glutathione/glutathione disulfide (GSH/GSSG) ratio in Zucker diabetic fatty (ZDF) rats [58]. Within the IMS, O2•- produced by NOX4 must be detoxified by SOD1. Otherwise, it exerts damage to the inner and outer mitochondrial membrane [59]. It will be a matter of future research to clarify whether diabetes-induced NOX4 upregulation is sufficient to rupture the outer membrane, initiate the mPTP, or induce mitochondrial outer membrane permeabilization (MOMP), and thereby trigger the redistribution of the IMS cell death proteins. Indeed, it has been demonstrated that NOX4 upregulation mediates TGF-β-induced apoptosis in podocytes, although it was not investigated whether IMS cell death proteins were engaged in this process [60].
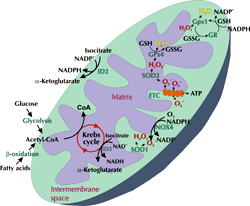
Investigations have also focused on deficiencies in the antioxidant network within the mitochondria, including coenzyme Q, SOD2, and GPx4 [34]. The consequence of an imbalance in these ROS generators and antioxidants within the diabetic renal cortex is an elevation in oxidative stress within the mitochondria, which can trigger the intrinsic cell death cascade [61]. Figure 1 summarizes the key players involved in redox signaling in the mitochondria. Under diabetic conditions, this redox signaling is disrupted, resulting in increased oxidative stress.
 |
 |
Figure 1. Redox signaling within mitochondria. ROS in the form of superoxide (O2.-) can be produced via the electron transport chain (ETC) or NOX4 within the inner mitochondrial membrane (IMM). O2.- is released from complexes I and III of the ETC into the matrix or intermembrane space. O2.- released from NOX4 or complex III enters the intermembrane space where it is dismutased by SOD1 into hydrogen peroxide (H2O2). O2.- within the matrix is converted into H2O2 by SOD2. In turn, H2O2 is detoxified into H2O via GPx1 in the intermembrane space or via GPx4 within the matrix. This delicate balance of ROS within the mitochondria is dependent upon the Krebs (citric acid) cycle, the conversion of NAD(P)+ to NAD(P)H, and vice versa. Under diabetic conditions this redox signaling is disrupted, resulting in increased oxidative stress. |
|
Finally, it should be mentioned that results from studies on the occurrence of mitochondrial ROS/NS in diabetes and the amounts produced are inconsistent to some extent. While it is now generally accepted that mitochondria-derived ROS/NS are increased in diabetes, some studies have reported an overall decrease [62, 63]. The discrepancy may result from the cellular heterogeneity and differing metabolic responses of the cells in the kidney [64]. Further research is necessary to clarify this issue.
4. Consequences of oxidative stress in the diabetic kidney
4.1 Nuclear DNA damage
Oxidative stress is known to contribute to a range of harmful intracellular events, including DNA damage within the nucleus. This damage may ultimately cause dysfunction, cell death, and long-term risk of oncogenesis. Generally, oxidative DNA damage, in the form of oxidized bases, occurs as frequently as one million times per day in the human body as a consequence of normal metabolism and other environmental factors [65, 66]. Cells rely on high energy DNA repair mechanisms to clear DNA damage. This includes the recruitment of the base excision repair (BER) protein complex, amongst others, to repair this type of damage [67]. The formation of general DNA repair complexes (e.g. ataxia telangiectasia mutated (ATM), ataxia and rad-related kinase (ATR)) involves the activation of proteins that target gene expression and protein production to respond rapidly to detected damage. Two of the more significant transcription factors are p53 and NF-κB, both of which regulate gene expression.
In cases where the DNA damage level is high, the recruitment of large volumes of repair complexes can result in rapid depletion of available ATP due to elevated energy demand, e.g. by cleavage of the damage-sensing protein poly (ADP-ribose) polymerase (PARP) [68, 69]. This creates the potential for unregulated cell death (i.e. necrosis) as there is insufficient energy for cells to undergo ATP-dependent programmed cell death (apoptosis) [70]. This "switch" in DNA damage-related cell death PARP-1 activation has been observed in canine renal cells [71, 72]. However, if the DNA damage by ROS is sub-lethal, cells can sufficiently repair the damage and even adapt to chronic change [73].
Mitochondria are involved in sensing nuclear DNA damage. They are able to influence cellular responses, including the release of apoptosis-inducing factor (AIF), a protein involved in DNA fragmentation and chromatin condensation during apoptosis [74]. In the pathogenesis of diabetic nephropathy, oxidative stress-related damage to mitochondria contributes to apoptosis and loss of podocytes, which are key events in the progression of the disease [75].
The ability for cells to adapt to increased levels of ROS/NS is a critical component in homeostatic protection. Due to the frequency of oxidative DNA damage across the body, cells have robust mechanisms to identify and repair this damage to prevent cumulative genotoxicity or mutations. BER is one of the mechanisms employed by mammalian cells to repair DNA damage caused by oxidation. [76, 77]. In normal cells, the BER pathway is regulated by 3 main proteins: OGG1, MTH1, and MUTYH that constitute a part of the BER repair complex [78, 79]. When an oxidized deoxyguanosine base (8-oxo-dG) is repaired by BER, the dissected base (8-OHdG) is discharged from the cell and filtered through the kidney [80].
Recent evaluations of DNA breaks induced by oxidative stress showed that they are increased in patients with type 1 and type 2 diabetes, although most studies involved analyses of peripheral lymphocytes [81-83]. The same phenomenon has also been observed in gestational diabetes [84, 85]. Of particular interest is the association with renal complications. The direct effect of high glucose on ROS/NS production and subsequent DNA damage has been assessed in rat models of type 1 diabetes [86]. These studies support the hypothesis that oxidative stress-induced damage occurs in the kidneys of diabetic patients. Clinical investigation into the effects of oxidative stress-induced DNA damage on renal cells is limited, in part because of the need for renal biopsies. However, it is possible to determine indirectly the level of oxidative damage in a patient by analyzing the urinary excretion of oxidized DNA bases in the form of 8-OHdG. This validated method has revealed increased excretion rates in patients with diabetes, corresponding to an increase in generalized oxidative stress-induced DNA damage throughout the body [80].
Interestingly, the occurrence of 8-oxo-dG base damage has been found to coincide with diabetic complications and may be a predictive biomarker for detecting the severity of diabetic complications, including retinopathy and nephropathy [80, 87, 88]. Although a higher excretion rate is correlated with severity of diabetic nephropathy, this critical biomarker can give an idea of systemic DNA damage only (i.e. relative damage in the entire body), but not the specific incidence in an individual organ. Another limiting factor for the use of 8-OHdG as biomarker for renal damage is that the excretion of 8-OHdG is affected by other compounding factors, including exercise and environmental toxins [89-91]. This limits the clinical implications of these studies as an individual's baseline excretion rate can vary significantly.
4.2 Mitochondrial DNA damage
In addition to the effects of nucleic DNA damage, mtDNA in the diabetic kidney is also susceptible to damage by ROS/NS [18]. The close proximity of mtDNA to the ETC within the mitochondrial matrix makes this type of DNA highly susceptible to damage by ROS/NS produced during OXPHOS. This mechanism is generally considered to account for the increased observation of oxidized mtDNA compared with nuclear DNA damage [92]. Although mtDNA damage is ultimately repaired after a short insult, longer insults lead to persistent damage that could subsequently result in apoptosis [93]. MtDNA damage is also repaired through the BER pathway by the excision of 8-OHdG, although there seems to be some slight differences between the recruited proteins in each organelle. For example, nucleic DNA is repaired primarily by DNA ligase I, whilst mtDNA damage is repaired by DNA ligase III [94].
The role of oxidative stress in DNA damage and mitochondrial dysfunction has already been established in other pathologies such as in neurodegeneration, heart failure, and diabetic retinopathy [95-97]. In a recent clinical study, a link was found between diabetes, damaged mtDNA, and functionally altered mtDNA. Indeed, changes in mtDNA preceded bioenergetic dysfunction, leading the authors to propose that systemic mitochondrial dysfunction initiated by glucose-induced mtDNA damage may be involved in the development of diabetic nephropathy [98].
4.3 Compromised DNA repair mechanisms
Susceptibility to oxidative stress-induced DNA damage has been shown to correlate with certain gene polymorphisms in the BER pathway in type 2 diabetes complications such as distal symmetric polyneuropathy [99, 100]. One study examined changes in the gene expression of the BER complex protein MUTYH, an important protein in the response to cellular stress, following treatment with TGF-β1. The investigators found that expression of the protein in renal proximal tubule cells differs from that in interstitial cells [101]. Furthermore, oxidative DNA damage, as measured by 8-oxoG, was positively associated with renal fibrosis, suggesting that MUTYH mediates tubulointerstitial damage [101]. Other studies have indicated that suppression of MUTYH may actually be protective of renal disease [102, 103]. Importantly, polymorphisms found in these DNA repair pathway genes have also been shown to increase the risk of ESRD progression in a cohort of mixed-cause patients undergoing hemodialysis [102]. It would be interesting to examine this effect in the context of diabetic nephropathy, and earlier in the progression of the disease, to determine whether this correlates with patient risk of progression to ESRD.
Besides BER, additional repair pathways such as homologous recombination and mismatch repair respond to critical DNA damage [104]. When DNA repair fails to proceed adequately, there is a danger of cell death by necrosis due to the high energy requirements of the DNA repair processes [70]. Thus, DNA repair proteins may be inactivated where possible to downregulate their functional activity for the salvation of apoptotic pathways [104]. However, loss of important repair processes, whether mediated by oxidative stress itself or by other factors, is thought to compromise the integrity of mitochondrial and nuclear DNA. Further investigation is required to find out how the repair mechanisms are compromised in diabetic nephropathy.
5. Modes of cell death during diabetic renal disease
Cell death is a natural consequence of oxidative stress when damage exceeds the ability of the cell to respond and repair the insult. The two main types of cell death observed in diabetic nephropathy are apoptosis and necrosis. Apoptosis is a controlled process that is highly dependent on the continued production of ATP to fuel this high-energy process [69]; it is characterized by cell shrinkage, chromatin condensation, and DNA fragmentation [61]. Apoptosis occurs through two types of pathways. One is the death receptor pathway (extrinsic apoptotic pathway) that includes FAS/FAS-ligand; the second is the mitochondrial pathway (intrinsic apoptotic pathway) that requires regulatory proteins such as the Bcl2-related family [105]. Necrosis, by comparison, is considered uncontrolled and catastrophic. In necrotic cell death, ATP production is insufficient to meet the energy demands of the cell, leading to rupture of cellular contents [106]. This generates a high-level inflammatory response, which is not observed in apoptosis. Each type of cell death has physiological consequences that correspond to the clinical presentation of a disease. Finally, ER stress [107] and cellular senescence within key cell types of the kidney [108] are two additional mechanisms that are now regarded as contributing to cell death processes in diabetic nephropathy.
5.1 Induction of apoptosis
In diabetic nephropathy, pathogenic lesions are characterized by initial hypertrophy followed by a gradual loss of renal mass, sclerosis, and fibrosis. Apoptosis is known to contribute to the later process in both humans and animals [109-111]; it includes the upregulation of cell surface markers which encourages phagocytosis (removal) by resident and circulating immune cells. These surface markers include CD36, CD95 (FAS-ligand), CD74 (MIF-R), OPG, and TRAIL, which are increased in diabetic kidneys [112-115]. FAS-ligand binds to FAS. It has been shown that plasma FAS levels are elevated in diabetes, and that the rate of apoptosis begins early in the development of proteinuria in renal cells [116, 117]. One study in diabetic female rats found that an increase in FAS/FAS-ligand could be attenuated by blockade of the renin-angiotensin system (RAS) [118]. The inner cell membrane portion of the FAS-ligand includes a FAS-associated protein with death domain (FADD). It has been shown that FADD and its dimeric counterpart FADD-DD play an important role in mediating cell death in renal tubule cells in the context of acute renal injury, and that they can mediate early diabetic renal injury [119, 120]. However, it is clear that additional pathways are involved since increased FAS-ligand levels do not directly correlate with apoptosis [116, 117].
Another event that may occur in diabetic nephropathy includes changes in internal pro-apoptotic proteins such as BASP1, Bcl-2 family proteins, and p53 [109]. The Bcl-2 family of proteins are highly conserved in diabetic nephropathy, and are involved in the regulation of both pro-survival and pro-apoptotic signaling. The numerous family members interact downstream of p53 and NF-κB to regulate cell survival. The anti-apoptotic protein Bcl-2 has been shown to play an important role in diabetic nephropathy [121] as it antagonizes the pro-apoptotic Bax and Bak proteins that together control mitochondrial cell death. An increase in the pro-apoptotic Bax relative to the anti-apoptotic Bcl-2 establishes a more pro-apoptotic phenotype; this ratio is a commonly used technique to establish the susceptibility of cells to apoptotic cell death. This change in ratio has been demonstrated in a murine model of type 2 diabetic nephropathy [122]. The increase in the observed pro-apoptotic Bax/Bcl-2 ratio could be reversed through blockade of the RAS pathway in mice overexpressing angiotensinogen in renal proximal tubular cells [123].
In human diabetic nephropathy, the data are limited regarding changes in Bcl-2 and Bax expression. However, it has been identified as a contributing factor in other forms of renal disease [124, 125]. Additionally, other Bcl-2 family members may also be linked with diabetic nephropathy. For example, overexpression of Bcl-2 modifying factor (BMF) is involved in apoptosis in both animal and human studies [126]. BMF is a BH3 only protein (a sub-class of smaller proteins within the Bcl-2 family) that indirectly activates Bax and Bak by inhibiting the related proteins Bid, Noxa, and Bik to promote cell death [127]. Other pathways implicated in diabetic nephropathy include Akt and p38 MAPK [128].
In the diabetic kidney, it is generally considered that ROS/NS-induced apoptosis is associated with specific changes observed in the disease, in particular tubulointerstitial changes [129]. For example, damage by high-glucose-induced ROS/NS has been associated with PKCdelta that regulates the cellular response protein p66Shc [130]. P66Sch mediates high-glucose and ANG II responses to oxidative stress and renal proximal tubule cell (PTC) injury via mitochondrial dysfunction and apoptosis pathways [131]. A recent review on renal tubular apoptosis in diabetic nephropathy reported that high-glucose is one of the major contributors to free radical- and ROS-induced cell death in these cells [61]. Further stressors relating to PTC injury include uric acid transport which is also associated with oxidative stress injury and apoptosis [132].
Prolonged oxidative stress is known to cause adaptive changes in cells. Cells that fail to adapt to the changes die by one of the many cell death pathways available to detect and respond to harmful cellular stress conditions. One pathway by which ROS/NS contributes to cell death is the classical apoptotic cascade engaging the Bcl-2 family proteins [133]. This pathway includes the activation of the Bax protein which translocates to the mitochondria of damaged cells and elicits changes in mitochondrial outer membrane permeability (MOMP) by the formation of the mitochondrial outer membrane pore. The downstream effects include the release of cytochrome c and subsequent caspase-3 cleavage.
Bax is activated by a range of transient protein interactions initiated by ROS/NS damage. However, in silico modeling also proposes a direct activation of Bax via oxidative interactions [134]. Bax is well known as part of the mitochondrial (intrinsic) pathway of apoptosis, although its full repertoire of cellular interactions is not yet understood. It has a clear role in p53-activated apoptosis, but evidence suggests the involvement of Bax in other forms of cell death as well [135, 136]. In apoptotic death, Bax undergoes a conformational change following activation which allows it to associate with mitochondrial membranes and to take part in the formation of the MOMP. In addition to Bax, the related protein Bak is also capable of forming these pores. Bak is a membrane-bound protein, whereas Bax is cytosolic in its inactive state. Studies show that Bax and Bak can oligomerize during MOMP formation [137]. While the precise morphology of this pore is still a matter of debate, it is hypothesized that there are two distinct pore types [138]. Bax expression has been found to be increased in a diabetic STZ rat model and in dbdb mice [122, 139]. However, expression in human renal cells is relatively unexplored in diabetic nephropathy.
Although necrotic cell death has also been observed in diabetic nephropathy [140], the full implications of its contribution to the disease are not yet understood.
5.2 Endoplasmic reticulum stress
Mitochondrial function is closely linked to endoplasmic reticulum (ER) function. ER stress is another important aspect of cellular pathology. It occurs simultaneously with oxidative stress in the context of the diabetic milieu and progression of diabetic complications [107, 141, 142]. In normal conditions, the ER is responsible for regulating protein folding. In situations of high demand or protein damage (as seen in oxidative stress), the unfolded protein response (UPR) is activated to restore ER homeostasis. This is mediated by the proteins XBP1 and IRE1, and allows for transient protection against damage [143, 144]. When the unfolded protein response is overwhelmed ER stress occurs. This is a cytotoxic process involving cellular dysfunction and the activation of proapoptotic factors such as IRE1 [144].
Cells with a high protein production rate are at an increased risk of cellular dysfunction by ER stress. Pancreatic beta-cells are an important example due to the continual production of proinsulin. In type 2 diabetes, this is particularly relevant as the development of insulin resistance and maximum insulin production places the pancreatic cells at an increased risk of ER stress [145, 146]. Additionally, proteinuria and hyperglycemia can directly induce ER stress in renal PTCs in vitro [147]. In fact, the upregulation of genes associated with the UPR positively associate with increased severity of diabetic nephropathy, which is regarded as a protective change [147]. ER stress has been shown to mediate renal pathology in diabetic nephropathy and to correspond with disease severity [148, 149]. Examples include albuminuria, which has been shown to cause ER stress by the induction of caspase-12 expression [150]. Furthermore, accumulation of protein in the proximal tubules is known to follow aldosterone administration in rat models (physiological elevated equivalent) and leads to PTC damage if not cleared by autophagy [151].
The ER is primarily responsible for regulating Ca2+. Oxidative stress has been found to alter Ca2+ homeostasis [152]. This alteration includes a release of Ca2+ from the ER into the cytosol, which in turn affects mitochondria and mitochondrial function [153]. In fact, calcium leakage has been shown to directly cause elevated ROS/NS production in mitochondria via interactions with OXPHOS [154]. Other proteins have been implicated in the reduction of elevated ROS/NS production via oxidative phosphorylation mechanisms in diabetes [155]. However, most of this research has focused on neurodegenerative or skeletal muscle models, not diabetic nephropathy.
In many disease processes, cell death by ER stress occurs via the mitochondrial apoptosis pathway [156]. In type 2 diabetes, ER stress appears to be upregulated and linked with an increase in both apoptosis and necrosis correlating with changes in inflammatory cytokine expression [140]. The translocation of Bax and Bak to the ER membrane may occur during ER stress-mediated apoptosis [157]. Furthermore, caspase-12 cleavage occurs downstream, indicating a pathway of cell death that is potentially independent of the mitochondria in human fibroblast cells [158]. In comparison, the upregulation and accumulation of another pro-apoptotic Bcl-2 family protein, BIM, at the ER membrane is associated with mitochondrial death pathways following caspase-12 activation [159, 160].
Bax/Bak oligomerization at the ER membrane followed by caspase-12 activation has also been demonstrated in mouse models [161]. However, murine caspase-12 is a homologue of human caspase-4. This variant has also been associated with cell death following ER stress [162]. Additionally, caspase-4 has been observed to mediate PTC death in some types of nephropathy [163], but is yet to be confirmed in diabetic kidney disease. Although human caspase-12 has been analyzed in many studies, its relevance to the general population has been questioned as the full homologue of the gene is only expressed in 2.8% of humans [164].
Additional caspases may be activated downstream of ER stress, including caspase-7 [158] and caspase-8 [165, 166]. It seems that the distribution of Bax to different organelles relates to the type of cell death induced [167]. The structure of the reported ER membrane pore is not yet known, but early results point to changes in membrane permeability [157].
Autophagy is another cell death pathway that has been observed when key components of the mitochondrial apoptotic pathway (i.e. Bax/Bak, caspase-9) are disrupted [165]. Although this aspect is of importance in the field of cancer research and drug resistance, in the context of diabetic nephropathy, it is interesting to consider the implications of altered mitochondrial function in this pathway, particularly as the link between mitochondria and ER relays important signal transfer during cell death [153]. Furthermore, Bcl-2 family proteins, Bax and Bak, have also been linked to this interaction by regulating Ca2+ ER homeostasis and efflux to the cytosol [168]. Bcl-2 family proteins play a major role in mediating the response of the ER and mitochondria to oxidative damage. The role of Bax in the progression from ER stress to apoptosis is an interesting area for exploration in diabetic nephropathy, considering the role of Bax in the mitochondria-dependent cell death pathways. Mediation of ER stress could potentially provide a therapeutic target for the prevention of renal disease.
5.3 Cellular senescence
Another potential outcome of oxidative stress is cellular senescence. This is likely to be due to a range of factors, including the inherent growth-inhibition response to DNA mutagenic stimuli. Senescence is a useful state induced by cells to protect from mutagenesis without depleting cell mass by apoptosis. Cells may persist indefinitely in cell cycle arrest. This forms an important physiological function in terminally differentiated tissues such as neurons and adult stem cells, which retain the ability to exit cell cycle arrest.
Senescence of cells can be triggered by oxidative stress. This has been observed in many human cell lines with fibroblasts being particularly susceptible [169-171]. Oxidative stress-induced senescence has also been demonstrated in normal renal cells, and identified as a contributing factor to diabetic nephropathy in animal models and type 2 diabetic patients [169, 172, 173]. The senescent response to oxidative stress can include an increase in mitochondrial mass and mtDNA copy number [174]. Although increased circulatory mtDNA has been detected in patients with type 2 diabetes [98, 175], it may be possible that senescent cells are more susceptible to the effects of ROS and NOS, instead of inducing a protective state. For example, renal podocytes exist as terminally differentiated cells functionally equivalent to senescent cells, but highly susceptible to oxidative stress. There is evidence to suggest that dedifferentiation of these cells occur in diabetes, indicating exit from cell cycle arrest [108] and resulting in podocyte loss. Podocyte loss, as measured by urinary excretion, is associated with early glomerular pathology in diabetic patients [176].
6. Mitochondrial permeability transition as a pathway to renal injury
Since the kidney relies on oxidative phosphorylation (OXPHOS) to provide the bulk requirements of ATP for tubular reabsorption [177], it is not surprising that mitochondrial homeostasis is essential for an optimally functioning kidney. Indeed, in experimental diabetic nephropathy or renal proximal tubule cells exposed to high glucose, mitochondrial ATP content [178] and production [179, 180] are depleted. Mitochondria are able to compensate for decreased cellular ATP production by fusion. This mechanism is an effective adaptive response, as previously observed in other organs [181]. Notably, changes in mitochondrial morphology within renal proximal tubules in both human [182] and animal models of early diabetic nephropathy [183] have long been identified as part of the disease, with mitochondrial enlargement and swelling. Diabetes-induced increases in mitochondria-derived ROS/NS have been shown to provoke the dynamic changes in mitochondrial shape [184]. Another stimulator of change in mitochondrial shape is the induction of mitochondrial permeability transition. This mechanism is independent of the mitochondrial dynamics machinery and oxidative stress, and has been historically detected by electron microscopy imaging showing mitochondrial swelling.
Mitochondrial permeability transition (mPT) is an abrupt increase in inner mitochondrial membrane (IMM) permeability that allows the passage of solutes with molecular masses of less than 1,500 Da. This event is caused by unlocking the mitochondrial permeability transition pore (mPTP), an evolutionary, highly conserved channel [185]. The unlocked pore allows for immediate dissipation of the mitochondrial transmembrane potential and influx of solutes, causing expansion of the matrix and mitochondrial swelling. The loss of the inner mitochondrial membrane potential and the inability to maintain a pH gradient due to proton influx also disrupts mitochondrial ATP synthesis, leading to energy depletion; persistent opening can induce necrotic cell death [186]. Sufficient swelling of the mitochondrial matrix may result in rupture of the outer mitochondrial membrane. This causes cytochrome c release with subsequent caspase activation, and results in cellular apoptosis [187]. Opening of the mPTP prevents mitochondria from generating ATP by OXPHOS, and allows reversal of the FoF1 ATP synthase, causing hydrolysis of the ATP produced by glycolysis or any remaining functional mitochondria [178].
The mPTP is composed of multiple macromolecular components which are not yet fully characterized [178, 180, 181]. It was initially thought that mPTP was comprised of the adenine nucleotide translocase (ANT), voltage-dependent anion channel (VDAC), and non-pore forming regulatory component cyclophilin D (CypD), the mitochondrial isoform of the peptidylprolyl cis-trans isomerase cyclophilin chaperone family [188, 189]. The mPTP was thought to be generated as one contiguous pore spanning the intermembrane space [190]. However, gene ablation studies suggested that VDAC and ANT play only a limited role in mPT [191-193]. Also, a significant role for CypD in mPT was established through the development of CypD-deficient mice (Ppif-/-) whose mitochondria do not undergo syclosporin A (CsA)-sensitive mPT [194]. CypD is considered to regulate mPT by facilitating a calcium-triggered conformational change in the mPTP, converting it into an open state [195]. CypD-deficient mice (Ppif-/-) are protected from ischemia/reperfusion injury of the heart and focal cerebral ischemia, implicating a role for CypD-dependent mPT opening and subsequent necrotic cell death in end-organ injury [194, 196]. Furthermore, mouse hepatocytes from CypD-deficient mice showed resistance to necrotic cell death induced by ROS and calcium overload [197]. In an updated model of the mPTP, dimers of the F1Fo ATP synthase form the mPTP within the IMM, with CypD binding to the lateral stalk of the FoF1 ATP synthase [198]. Further studies have identified the c-subunit ring of the Fo (i.e. ab2c10-14, Escherichia coli nomenclature) of the FoF1 ATP synthase to form the inner pore of the mPTP [199]. However, this model has not yet been validated.
Mitochondrial permeability transition is induced by mitochondrial sequestration of high levels of calcium, and is sensitized by factors such as decreased ATP levels, inorganic phosphate, and alkaline pH [178]. Hydrogen peroxide and other ROS may lead to induction of mPT [200]. mPT can also induce changes to the mitochondrial respiratory chain, including inhibition of complex I [201] and loss of cytochrome c [202]. Both of these conditions contribute to a reduced and decelerated flow of electrons through the respiratory chain, favoring the one-electron reduction of molecular oxygen. Finally, these events result in the generation of superoxide radicals [203, 204]. Loss of cytochrome c from the mitochondrial IMS reduces ATP synthesis, and further increases electron leak and ROS generation, setting up a feed-forward cycle of ROS-induced ROS generation [205]. A recent study has found that Bax mediates mPTP opening, which may cause ATP depletion and cellular necrosis [206].
Recently, a role for mPT has been implicated in renal injury in the kidney. Swelling of mitochondria within renal proximal tubules of the kidney has been demonstrated in early experimental diabetic nephropathy [183] and in kidneys from diabetic patients [182]. Ppif-/- mice are protected from acute ischemic renal injury [207]. Moreover, in a short-term model of streptozotocin (STZ)-induced diabetes, rats treated with cyclosporin A (CsA), an inhibitor of mPT, were protected against glomerular hypertrophy and extracellular matrix (ECM) accumulation [208]. A previous study has suggested that renal mitochondria from Goto-Kakizaki rats, a model of type 2 diabetes, had enhanced mPT. However, mPT was only measured indirectly by extramitochondrial calcium flux and its amelioration by CsA in vitro [209]. Recent studies from our laboratory have suggested that mPT is a candidate for mediating ROS-induced renal damage [179]. However, interruption in physiological mPT and the downstream consequences, including cell death, have not been extensively studied in models of diabetic nephropathy. It is possible that enhanced susceptibility to the mPT contributes to the development of diabetic nephropathy.
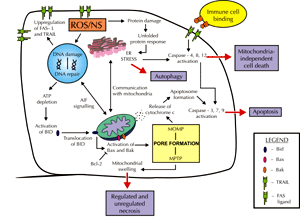
In summary, diabetes-induced oxidative stress is able to stimulate numerous cellular pathways that eventually lead to cell death. Different types of cell death may occur depending on the parts of the cell that are damaged, as illustrated in Figure 2. Apoptosis, mostly induced by MOMP formation, mitochondrial shrinkage, and cytochrome c release, activates caspases via intrinsic or extrinsic pathways. Necrosis induced by MPTP formation results in mitochondrial swelling and lysis. As highlighted in this review, evidence for these major cell death pathways can be found in the diabetic kidney.
 |
|
Figure 2. Lethal consequences of ROS/NS. The presence of high levels of ROS/NS can lead to downstream activation of cell death pathways. The type of cell death that occurs depends on the parts of the cell that are damaged. For example, oxidized protein damage can induce ER stress and mediate mitochondrial-independent cell death. However, crosstalk with the mitochondria can induce apoptosis via release of cytochrome c if suitable stimuli are present. DNA damage by ROS/NS can induce a range of signaling pathways, including the upregulation of gene expression of the cell surface markers FAS-L and TRAIL. These markers encourage cytotoxic immune cells to bind and activate the extrinsic apoptosis pathway. Furthermore, communication with mitochondria can lead to apoptotic or necrotic cell death if ATP is depleted. Activation of Bax and Bak can cause mitochondrial pore formation and opening, resulting in either mitochondrial swelling and necrosis or apoptosis via cytochrome c release. This process can be blocked by the anti-apoptotic protein BCl-2. The persistence of ROS/NS in diabetes may result in loss of cell mass and subsequent decrease in renal function. |
|
7. Targeting mitochondrial ROS in diabetic nephropathy
Despite increasing evidence supporting the role of mitochondrial oxidative stress in experimental diabetic nephropathy, the evidence from clinical trials using antioxidants has not confirmed a beneficial effect. For example in the Heart Outcomes Prevention Evaluation (HOPE) trial, vitamin E treatment for 4.5 years failed to confer benefits in cardiovascular outcomes or nephropathy [13]. Lack of specificity and failure to target the correct sites of ROS production are two reasons often mentioned for the potential failure of vitamin E in the clinic. Therefore, the idea of specifically targeting ROS within ROS-producing organelles such as the mitochondria has been investigated. This strategy aims to increase specificity and to avoid targeting important physiological ROS outside the mitochondria because such ROS may be required for cell signaling. Mitochondria-targeting antioxidants have therefore been suggested as a potential therapeutic strategy [210] for the prevention of cisplatin-induced nephropathy [211], acute kidney injury [212], CKD, and diabetic nephropathy. This strategy may also hold promise for the treatment of cardiovascular diseases [213].
The most effective antioxidants would be those that can cross the outer and inner mitochondrial membrane. Therefore, antioxidants were conjugated with a positively charged triphenylphosphonium cation (TPP+) to yield compounds such as quinone analogues, mitoquinone (Mito Q), SkQ1 and SkQR1, as well as mitovitamin E, mitophenyltertbutyline, and the SOD mimetic Mito-CP [212]. These antioxidants accumulate within the mitochondrial matrix at concentrations several-fold higher than within the cytosolic compartment due to the high negative membrane potential of the IMM. A study by Chacko et al. in type 1 diabetic Ins2-Akita mice showed that oral administration of MitoQ for 12 weeks prevented diabetic kidney damage [214]. Interstitial fibrosis and glomerular damage were significantly reduced in the treated animals. MitoQ also reduced the expression of the pro-fibrotic transcription factors phospho-Smad2 and 3, and prevented the nuclear accumulation of β-catenin, a member of the Wnt pathway that has been implicated in pathological processes such as fibrosis. Similarly, Mito-CP, the SOD mimetic composed of a lipophilic cationic nitroxide conjugated to TPP+, prevented cisplatin-induced renal injury by effectively limiting oxidative and nitrosative stress, preventing mitochondrial structural damage and tubular injury, attenuating renal inflammation, preventing renal dysfunction, and reducing renal cell death in 6-8 week-old male C57Bl/6J mice [211].
Other strategies to increase the antioxidant defense in the mitochondria include overexpression of hemeoxygenase-1 (HO-1), an antioxidant enzyme that is not normally present in mitochondria [215]. To achieve this, a plasmid containing a mitochondrial permeability sequence was fused to the HO-1 gene sequence and transfected into HEK293 renal epithelial cells. As a result, Mito-HO-1-expressing cells were protected from hypoxia-dependent cell death and loss of mitochondrial membrane potential. However, the treatment was afflicted with long-term limitations due to the negative impact on heme-containing mitochondrial proteins.
Ebselen is a synthetic selenocyteine-containing mimetic of the cytosolic and mitochondrial enzyme glutathione peroxidase-1 (GPx1) [216], In our type 1 diabetic ApoE knockout mouse model, ebselen had renoprotective effects, with reductions in renal inflammation and fibrosis [21, 217]. Ebselen has been shown to cross into the mitochondrial matrix without the need for a leader sequence. It is activated by the intramitochondrial glutathione and thioredoxin systems, making this an extremely attractive therapy for diabetic nephropathy [218]. In summary, these results support the hypothesis that mitochondrially targeted therapies may be beneficial in the treatment of diabetic nephropathy.
An alternative approach has been the development of mitochondrially targeted Szeto-Schiller (SS) tetrapeptides that bind to cardiolipin on the IMM. The SS tetrapeptide/cardiolipin complex protects cardiolipin from peroxidative damage by cytochrome c, thus protecting mitochondrial crista and preserving mitochondrial structure and function. Use of the SS-31 peptide, which is known to scavenge ROS and inhibit mPTP opening, protects mitochondrial structure and function. In a rat model of ischemic kidney injury, it accelerated recovery of ATP, reduced apoptosis and necrosis of tubular cells, and abrogated tubular dysfunction [219]. Furthermore, SS-31 peptide decreased ischemia/reperfusion-mediated oxidative stress and the inflammatory response in tubular cells. The SS peptide known as Bendavia (Stealth Peptides, Newton Centre, Massachusetts) has advanced into the clinic as a phase 2a randomized controlled trial (EMBRACE STEMI) to evaluate its safety, tolerability, and efficacy on reperfusion injury in patients after myocardial infarction [220]. A second clinical trial will evaluate the efficacy of Bendavia on renal parameters (estimated glomerular filtration rate (eGFR), renal volume and perfusion, inflammation and urinary osmolarity). This is a promising strategy for the restoration of mitochondrial injury, but needs first to be tested preclinically in models of diabetic nephropathy.
The strategy of specifically targeting the mPTP has seen the development of mPTP-opening inhibitors such as cyclosporin A (CsA) or sanglifehrin A [221]. CsA is a well-known immunosuppressant and inhibitor of mPT; it acts via inhibition of peptidyl-prolyl cis-trans isomerase (PPIase) activity of cyclophilin D [222, 223]. Clinically, CsA has already shown benefits in reducing human cardiac ischemia/reperfusion injury [224]. However, CsA is nephrotoxic [225], and interacts with the calcineurin pathway, which is activated in diabetic nephropathy [208]. These adverse properties limit its application in the treatment of diabetic nephropathy. Analogues of CsA have been generated, including Debio-025 (Alisporivir). Debio-025 is non-immunosuppressive [226], and unlike CsA, it does not display affinity for calcineurin [227], but it selectively inhibits cyclophilin D, prevents cell death [228, 229], and restores mitochondrial function [230]. To date, Debio-025 has not been tested in experimental models of diabetes.
MitoTempo is a mitochondrially-targeted SOD1. In response to excessive ROS production and calcium overload, it prevents mPTP opening in cultured proximal tubular epithelial cells [231]. Moreover, a recent study targeting glycogen synthase kinase (GSK) 3β through the use of 4-benzyl-2-methyl-1,2,4-thiadiazolidine-3,5-dione (TDZD-8), a highly selective small-molecule inhibitor of GSK3β, has shown promise in protecting podocytes against mPTP opening, ROS production, and apoptosis induced by adriamycin [232].
Other treatments have improved diabetic nephropathy end-points by increasing antioxidant capacity through upregulation of antioxidant response genes using Nrf2 activators such as bardoxolone methyl [233, 234], curcumin [235], sulforaphane [236], cinnamic aldehyde [236], resveratrol [237], and ebselen [21]. Recent studies have shown that metformin, an oral hypoglycemic drug, possesses antioxidant properties, and is effective in lowering end-points of diabetic nephropathy in a rat model [238]. Other studies have investigated the targeting of mitophagy pathways in the hope of restricting the accumulation of fragmented mitochondria in the renal cortex, a process shown to be linked with the development of diabetic nephropathy [34]. Furthermore, some treatments, not necessarily targeted at mitochondria, have included mangiferin, a natural C-glucosyl xanthone and polyhydroxy polyphenol compound that has shown protection against diabetic nephropathy in STZ-induced diabetic Wistar rats via different mechanisms, including inhibition of the PKC, MAPKs (p38, JNK and ERK1/2), NF-κB, and TGF-β1 pathways [239].
Finally, the specific targeting of the main enzymatic source of ROS/NS in the kidney, NOX4, with specific NOX1/NOX4 inhibitors has shown tremendous potential for the treatment of diabetic nephropathy [240], as discussed earlier in section 3. It is unknown whether these inhibitors enter the mitochondria per se, but recent evidence suggests that they mediate their inhibitory effect on NOX4, and thus they impact diabetic nephropathy via pathways that include enzymes located within the mitochondria [56]. The existence of disparate NOX inhibitors such as the pan-NOX inhibitors, VAS2870 and VAS3947 [241], await further preclinical analysis with respect to their effectiveness in preventing or reversing diabetic nephropathy. The more specific NOX1/NOX4 inhibitor, GKT137831, which is developed by Genkyotex, has completed phase 1 clinical trials with encouraging results regarding safety and tolerability. A phase 2 clinical study is now planned in diabetic nephropathy patients [242].
8. Concluding remarks and future perspective
Diabetic nephropathy is a leading cause of ESRD. Despite significant advances in the understanding of the cellular mechanisms that are responsible for its initiation and progression, it remains therapeutically elusive. It is clear that blood glucose and blood pressure control are insufficient to prevent its progression, and that new, more effective treatment options are urgently needed. The therapeutic targeting of diabetic kidney mitochondria is a relatively new area that has gained attention due to the involvement of mitochondria in cell death pathways such as apoptosis and necrosis implicated in key cell types such as podocytes.
The involvement of oxidative and nitrosative stress in mediating many of the observed changes has focused attention on antioxidants that specifically target mitochondria. Several approaches have been attempted, as outlined in this review, with a more nuanced approach required to restore redox homeostasis rather than depletion of important oxidants required for signaling. The interwoven involvement of ER stress with oxidative stress means that new targets may be found by closer examination of ER stress-driven pathways.
Preventing the consequences of increased ROS/NS on nuclear and mitochondrial DNA damage via enhanced surveillance by DNA repair mechanisms may offer an alternative solution. Preventing mPT pore formation is another new area with the potential to regulate renal injury. It is only by careful analysis of the cellular mechanisms governing diabetic renal injury that novel targets will be recognized and verified preclinically prior to translation into the clinic. These approaches promise to pave the way for new, more effective treatment options for diabetic nephropathy.
Disclosures: The authors report no conflict of interests.
Acknowledgments:
This work has been supported in part by the Victorian Government's OIS Program.
References
- Zimmet P, Alberti KG, Shaw J. Global and societal implications of the diabetes epidemic. Nature 2001. 414(6865):782-787. [DOD] [CrossRef]
- Spiegel AM, Nabel EG. NIH research on obesity and type 2 diabetes: providing the scientific evidence base for actions to improve health. Nat Med 2006. 12(1):67-69. [DOD] [CrossRef]
- Shaw JE, Sicree RA, Zimmet PZ. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res Clin Pract 2010. 87(1):4-14. [DOD] [CrossRef]
- Sarnak MJ, Levey AS, Schoolwerth AC, Coresh J, Culleton B, Hamm LL, McCullough PA, Kasiske BL, Kelepouris E, Klag MJ, et al. Kidney disease as a risk factor for development of cardiovascular disease: a statement from the American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. Hypertension 2003. 42(5):1050-1065. [DOD] [CrossRef]
- Schiele F. Renal dysfunction and coronary disease: a high-risk combination. J Nephrol 2009. 22(1):39-45. [DOD]
- Dinneen SF, Gerstein HC. The association of microalbuminuria and mortality in non-insulin-dependent diabetes mellitus. A systematic overview of the literature. Arch Intern Med 1997. 157(13):1413-1418. [DOD] [CrossRef]
- Gerstein HC, Mann JF, Yi Q, Zinman B, Dinneen SF, Hoogwerf B, Halle JP, Young J, Rashkow A, Joyce C, et al. Albuminuria and risk of cardiovascular events, death, and heart failure in diabetic and nondiabetic individuals. JAMA 2001. 286(4):421-426. [DOD] [CrossRef]
- Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal 2014. 20(7):1126-1167. [DOD] [CrossRef]
- Jha JC, Gray SP, Barit D, Okabe J, El-Osta A, Namikoshi T, Thallas-Bonke V, Wingler K, Szyndralewiez C, Heitz F, et al. Genetic targeting or pharmacologic inhibition of NADPH oxidase nox4 provides renoprotection in long-term diabetic nephropathy. J Am Soc Nephrol 2014. 25(6):1237-1254. [DOD] [CrossRef]
- Tonelli M, Sacks F, Pfeffer M, Jhangri GS, Curhan G, Cholesterol and Recurrent Events (CARE) Trial Investigators. Biomarkers of inflammation and progression of chronic kidney disease. Kidney Int 2005. 68(1):237-245. [DOD] [CrossRef]
- Okamura DM, Himmelfarb J. Tipping the redox balance of oxidative stress in fibrogenic pathways in chronic kidney disease. Pediatr Nephrol 2009. 24(12):2309-2319. [DOD] [CrossRef]
- McQueen MJ, Lonn E, Gerstein HC, Bosch J, Yusuf S. The HOPE (Heart Outcomes Prevention Evaluation) Study and its consequences. Scand J Clin Lab Invest Suppl 2005. 240:143-156. [DOD] [CrossRef]
- Lonn E, Yusuf S, Hoogwerf B, Pogue J, Yi Q, Zinman B, Bosch J, Dagenais G, Mann JF, Gerstein HC. Effects of vitamin E on cardiovascular and microvascular outcomes in high-risk patients with diabetes: results of the HOPE study and MICRO-HOPE substudy. Diabetes Care 2002. 25(11):1919-1927. [DOD] [CrossRef]
- Marchioli R, Levantesi G, Macchia A, Marfisi RM, Nicolosi GL, Tavazzi L, Tognoni G, Valagussa F. Vitamin E increases the risk of developing heart failure after myocardial infarction: Results from the GISSI-Prevenzione trial. J Cardiovasc Med (Hagerstown) 2006. 7(5):347-350. [DOD] [CrossRef]
- Hakim FA, Pflueger A. Role of oxidative stress in diabetic kidney disease. Med Sci Monit 2010. 16(2):RA37-RA48. [DOD]
- Onozato ML, Tojo A, Goto A, Fujita T, Wilcox CS. Oxidative stress and nitric oxide synthase in rat diabetic nephropathy: effects of ACEI and ARB. Kidney Int 2002. 61(1):186-194. [DOD] [CrossRef]
- Chander PN, Gealekman O, Brodsky SV, Elitok S, Tojo A, Crabtree M, Gross SS, Goligorsky MS. Nephropathy in Zucker diabetic fat rat is associated with oxidative and nitrosative stress: prevention by chronic therapy with a peroxynitrite scavenger ebselen. J Am Soc Nephrol 2004. 15(9):2391-2403. [DOD] [CrossRef]
- Kakimoto M, Inoguchi T, Sonta T, Yu HY, Imamura M, Etoh T, Hashimoto T, Nawata H. Accumulation of 8-hydroxy-2'-deoxyguanosine and mitochondrial DNA deletion in kidney of diabetic rats. Diabetes 2002. 51(5):1588-1595. [DOD] [CrossRef]
- Ha H, Lee HB. Reactive oxygen species amplify glucose signalling in renal cells cultured under high glucose and in diabetic kidney. Nephrology (Carlton) 2005. 10(Suppl):S7-S10. [DOD] [CrossRef]
- Lee HB, Yu MR, Yang Y, Jiang Z, Ha H. Reactive oxygen species-regulated signaling pathways in diabetic nephropathy. J Am Soc Nephrol 2003. 14(8 Suppl 3):S241-S245. [DOD]
- Chew P, Yuen DY, Stefanovic N, Pete J, Coughlan MT, Jandeleit-Dahm KA, Thomas MC, Rosenfeldt F, Cooper ME, de Haan JB. Antiatherosclerotic and renoprotective effects of ebselen in the diabetic apolipoprotein E/GPx1-double knockout mouse. Diabetes 2010. 59(12):3198-3207. [DOD] [CrossRef]
- Xiao H, Li Y, Qi J, Wang H, Liu K. Peroxynitrite plays a key role in glomerular lesions in diabetic rats. J Nephrol 2009. 22(6):800-808. [DOD]
- Gorin Y, Block K, Hernandez J, Bhandari B, Wagner B, Barnes JL, Abboud HE. Nox4 NAD(P)H oxidase mediates hypertrophy and fibronectin expression in the diabetic kidney. J Biol Chem 2005. 280(47):39616-39626. [DOD] [CrossRef]
- Kanauchi M, Nishioka H, Hashimoto T. Oxidative DNA damage and tubulointerstitial injury in diabetic nephropathy. Nephron 2002. 91(2):327-329. [DOD] [CrossRef]
- Hinokio Y, Suzuki S, Hirai M, Suzuki C, Suzuki M, Toyota T. Urinary excretion of 8-oxo-7, 8-dihydro-2'-deoxyguanosine as a predictor of the development of diabetic nephropathy. Diabetologia 2002. 45(6):877-882. [DOD] [CrossRef]
- Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocr Rev 2002. 23(5):599-622. [DOD] [CrossRef]
- Studer RK, Craven PA, DeRubertis FR. Antioxidant inhibition of protein kinase C-signaled increases in transforming growth factor-beta in mesangial cells. Metabolism 1997. 46(8):918-925. [DOD] [CrossRef]
- Ha H, Lee HB. Reactive oxygen species as glucose signaling molecules in mesangial cells cultured under high glucose. Kidney Int Suppl 2000. 77:S19-S25. [DOD] [CrossRef]
- Border WA, Noble NA. TGF-beta in kidney fibrosis: a target for gene therapy. Kidney Int 1997. 51(5):1388-1396. [DOD] [CrossRef]
- Yamamoto T, Noble NA, Miller DE, Border WA. Sustained expression of TGF-beta 1 underlies development of progressive kidney fibrosis. Kidney Int 1994. 45(3):916-927. [DOD] [CrossRef]
- Ha H, Yu MR, Choi YJ, Kitamura M, Lee HB. Role of high glucose-induced nuclear factor-kappaB activation in monocyte chemoattractant protein-1 expression by mesangial cells. J Am Soc Nephrol 2002. 13(4):894-902. [DOD]
- Rhyu DY, Yang Y, Ha H, Lee GT, Song JS, Uh ST, Lee HB. Role of reactive oxygen species in TGF-beta1-induced mitogen-activated protein kinase activation and epithelial-mesenchymal transition in renal tubular epithelial cells. J Am Soc Nephrol 2005. 16(3):667-675. [DOD] [CrossRef]
- Forbes JM, Coughlan MT, Cooper ME. Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes 2008. 57(6):1446-1454. [DOD] [CrossRef]
- Higgins GC, Coughlan MT. Mitochondrial dysfunction and mitophagy: the beginning and end to diabetic nephropathy? Br J Pharmacol 2014. 171(8):1917-1942. [DOD]
- Dorn GW 2nd. Molecular mechanisms that differentiate apoptosis from programmed necrosis. Toxicol Pathol 2013. 41(2):227-234. [DOD] [CrossRef]
- Kyte J. Immunoferritin determination of the distribution of (Na+ + K+) ATPase over the plasma membranes of renal convoluted tubules. II. Proximal segment. J Cell Biol 1976. 68(2):304-318. [DOD] [CrossRef]
- Kyte J. Immunoferritin determination of the distribution of (Na+ + K+) ATPase over the plasma membranes of renal convoluted tubules. I. Distal segment. J Cell Biol 1976. 68(2):287-303. [DOD] [CrossRef]
- Agus ZS, Chiu PJ, Goldberg M. Regulation of urinary calcium excretion in the rat. Am J Physiol 1977. 232(6):F545-F549. [DOD]
- Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria: central role of complex III. J Biol Chem 2003. 278(38):36027-36031. [DOD] [CrossRef]
- St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem 2002. 277(47):44784-44790. [DOD] [CrossRef]
- Forbes JM, Ke BX, Nguyen TV, Henstridge DC, Penfold SA, Laskowski A, Sourris KC, Groschner LN, Cooper ME, Thorburn DR, Coughlan MT. Deficiency in mitochondrial complex I activity due to Ndufs6 gene trap insertion induces renal disease. Antioxid Redox Signal 2013. 19(4):331-343. [DOD] [CrossRef]
- Maessen DE, Stehouwer CD, Schalkwijk CG. The role of methylglyoxal and the glyoxalase system in diabetes and other age-related diseases. Clin Sci (Lond) 2015. 128(12):839-861. [DOD] [CrossRef]
- Gugliucci A, Menini T. The axis AGE-RAGE-soluble RAGE and oxidative stress in chronic kidney disease. Adv Exp Med Biol 2014. 824:191-208. [DOD]
- Coughlan MT, Thorburn DR, Penfold SA, Laskowski A, Harcourt BE, Sourris KC, Tan AL, Fukami K, Thallas-Bonke V, Nawroth PP, et al. RAGE-induced cytosolic ROS promote mitochondrial superoxide generation in diabetes. J Am Soc Nephrol 2009. 20(4):742-752. [DOD] [CrossRef]
- Rosca MG, Mustata TG, Kinter MT, Ozdemir AM, Kern TS, Szweda LI, Brownlee M, Monnier VM, Weiss MF. Glycation of mitochondrial proteins from diabetic rat kidney is associated with excess superoxide formation. Am J Physiol Renal Physiol 2005. 289(2):F420-F430. [DOD] [CrossRef]
- Kim KM, Kim YS, Jung DH, Lee J, Kim JS. Increased glyoxalase I levels inhibit accumulation of oxidative stress and an advanced glycation end product in mouse mesangial cells cultured in high glucose. Exp Cell Res 2012. 318(2):152-159. [DOD] [CrossRef]
- Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001. 414(6865):813-820. [DOD] [CrossRef]
- Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000. 404(6779):787-790. [DOD] [CrossRef]
- Kiritoshi S, Nishikawa T, Sonoda K, Kukidome D, Senokuchi T, Matsuo T, Matsumura T, Tokunaga H, Brownlee M, Araki E. Reactive oxygen species from mitochondria induce cyclooxygenase-2 gene expression in human mesangial cells: potential role in diabetic nephropathy. Diabetes 2003. 52(10):2570-2577. [DOD] [CrossRef]
- Zhan M, Usman IM, Sun L, Kanwar YS. Disruption of renal tubular mitochondrial quality control by Myo-inositol oxygenase in diabetic kidney disease. J Am Soc Nephrol 2015. 26(6):1304-1321. [DOD] [CrossRef]
- Bock F, Shahzad K, Wang H, Stoyanov S, Wolter J, Dong W, Pelicci PG, Kashif M, Ranjan S, Schmidt S, et al. Activated protein C ameliorates diabetic nephropathy by epigenetically inhibiting the redox enzyme p66Shc. Proc Natl Acad Sci U S A 2013. 110(2):648-653. [DOD] [CrossRef]
- Block K, Gorin Y, Abboud HE. Subcellular localization of Nox4 and regulation in diabetes. Proc Natl Acad Sci U S A 2009. 106(34):14385-14390. [DOD] [CrossRef]
- Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov 2011. 10(6):453-471. [DOD] [CrossRef]
- Dikalov SI, Dikalova AE, Bikineyeva AT, Schmidt HH, Harrison DG, Griendling KK. Distinct roles of Nox1 and Nox4 in basal and angiotensin II-stimulated superoxide and hydrogen peroxide production. Free Radic Biol Med 2008. 45(9):1340-1351. [DOD] [CrossRef]
- Jo SH, Son MK, Koh HJ, Lee SM, Song IH, Kim YO, Lee YS, Jeong KS, Kim WB, Park JW, et al. Control of mitochondrial redox balance and cellular defense against oxidative damage by mitochondrial NADP+-dependent isocitrate dehydrogenase. J Biol Chem 2001. 276(19):16168-16176. [DOD] [CrossRef]
- You YH, Quach T, Saito R, Pham J, Sharma K. Metabolomics reveals a key role for fumarate in mediating the effects of NADPH oxidase 4 in diabetic kidney disease. J Am Soc Nephrol 2015. In press. [DOD]
- Coughlan MT, Thallas-Bonke V, Pete J, Long DM, Gasser A, Tong DC, Arnstein M, Thorpe SR, Cooper ME, Forbes JM. Combination therapy with the advanced glycation end product cross-link breaker, alagebrium, and angiotensin converting enzyme inhibitors in diabetes: synergy or redundancy? Endocrinology 2007. 148(2):886-895. [DOD]
- Winiarska K, Focht D, Sierakowski B, Lewandowski K, Orlowska M, Usarek M. NADPH oxidase inhibitor, apocynin, improves renal glutathione status in Zucker diabetic fatty rats: a comparison with melatonin. Chem Biol Interact 2014. 218:12-19. [DOD] [CrossRef]
- Weisiger RA, Fridovich I. Mitochondrial superoxide simutase. Site of synthesis and intramitochondrial localization. J Biol Chem 1973. 248(13):4793-4796. [DOD]
- Das R, Xu S, Quan X, Nguyen TT, Kong ID, Chung CH, Lee EY, Cha SK, Park KS. Upregulation of mitochondrial Nox4 mediates TGF-beta-induced apoptosis in cultured mouse podocytes. Am J Physiol Renal Physiol 2014. 306(2):F155-F167. [DOD] [CrossRef]
- Habib SL. Diabetes and renal tubular cell apoptosis. World J Diabetes 2013. 4(2):27-30. [DOD] [CrossRef]
- Towler DA. Mitochondrial ROS deficiency and diabetic complications: AMP(K)-lifying the adaptation to hyperglycemia. J Clin Invest 2013. 123(11):4573-4576. [DOD] [CrossRef]
- Dugan LL, You YH, Ali SS, Diamond-Stanic M, Miyamoto S, DeCleves AE, Andreyev A, Quach T, Ly S, Shekhtman G, et al. AMPK dysregulation promotes diabetes-related reduction of superoxide and mitochondrial function. J Clin Invest 2013. 123(11):4888-4899. [DOD] [CrossRef]
- Nishikawa T, Brownlee M, Araki E. Mitochondrial reactive oxygen species in the pathogenesis of early diabetic nephropathy. J Diabetes Invest 2015. 6(2):137-139. [DOD] [CrossRef]
- Saul RL, Ames BN. Background levels of DNA damage in the population. Basic Life Sci 1986. 38:529-535. [DOD]
- Ames BN, Shigenaga MK. Oxidants are a major contributor to aging. Ann N Y Acad Sci 1992. 663:85-96. [DOD] [CrossRef]
- Dizdaroglu M. Base-excision repair of oxidative DNA damage by DNA glycosylases. Mutat Res 2005. 591(1-2):45-59. [DOD] [CrossRef]
- Feldenberg LR, Thevananther S, del Rio M, de Leon M, Devarajan P. Partial ATP depletion induces Fas- and caspase-mediated apoptosis in MDCK cells. Am J Physiol 1999. 276(6 Pt 2):F837-F846. [DOD]
- Eguchi Y, Srinivasan A, Tomaselli KJ, Shimizu S, Tsujimoto Y. ATP-dependent Steps in Apoptotic Signal Transduction. Cancer Res 1999. 59(9):2174-2181. [DOD]
- Leist M, Single B, Castoldi AF, Kuhnle S, Nicotera P. Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J Exp Med 1997. 185(8):1481-1486. [DOD] [CrossRef]
- Koh DW, Dawson TM, Dawson VL. Mediation of cell death by poly(ADP-ribose) polymerase-1. Pharmacol Res 2005. 52(1):5-14. [DOD] [CrossRef]
- Hassa PO. The molecular "Jekyll and Hyde" duality of PARP1 in cell death and cell survival. Front Biosci (Landmark Ed) 2009. 14:72-111. [DOD] [CrossRef]
- Laval J. Role of DNA repair enzymes in the cellular resistance to oxidative stress. Pathol Biol (Paris) 1996. 44(1):14-24. [DOD]
- Hong SJ, Dawson TM, Dawson VL. Nuclear and mitochondrial conversations in cell death: PARP-1 and AIF signaling. Trends Pharmacol Sci 2004. 25(5):259-264. [DOD] [CrossRef]
- Susztak K, Raff AC, Schiffer M, Bottinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes 2006. 55(1):225-233. [DOD] [CrossRef]
- Prakash A, Doublie S. Base Excision Repair in the Mitochondria. J Cell Biochem 2015. In press[DOD]
- Bohr VA, Stevnsner T, de Souza-Pinto NC. Mitochondrial DNA repair of oxidative damage in mammalian cells. Gene 2002. 286(1):127-134. [DOD] [CrossRef]
- Campalans A, Moritz E, Kortulewski T, Biard D, Epe B, Radicella JP. Interaction with OGG1 is required for efficient recruitment of XRCC1 to base excision repair and maintenance of genetic stability after exposure to oxidative stress. Mol Cell Biol 2015. 35(9):1648-1658. [DOD] [CrossRef]
- Nakabeppu Y, Tsuchimoto D, Ichinoe A, Ohno M, Ide Y, Hirano S, Yoshimura D, Tominaga Y, Furuichi M, Sakumi K. Biological significance of the defense mechanisms against oxidative damage in nucleic acids caused by reactive oxygen species: from mitochondria to nuclei. Ann N Y Acad Sci 2004. 1011:101-111. [DOD] [CrossRef]
- Wu LL, Chiou CC, Chang PY, Wu JT. Urinary 8-OHdG: a marker of oxidative stress to DNA and a risk factor for cancer, atherosclerosis and diabetics. Clin Chim Acta 2004. 339(1-2):1-9. [DOD] [CrossRef]
- Giovannini C, Piaggi S, Federico G, Scarpato R. High levels of gamma-H2AX foci and cell membrane oxidation in adolescents with type 1 diabetes. Mutat Res 2014. 770:128-135. [DOD] [CrossRef]
- Prasad M, Bronson SC, Warrier T, Badarinath A, Rai S, Baid K, Sitaraman S, George A, Moses A, Saraswathy R, et al. Evaluation of DNA damage in Type 2 diabetes mellitus patients with and without peripheral neuropathy: A study in South Indian population. J Nat Sci Biol Med 2015. 6(1):80-84. [DOD] [CrossRef]
- Cinkilic N, Kiyici S, Celikler S, Vatan O, Oz Gul O, Tuncel E, Bilaloglu R. Evaluation of chromosome aberrations, sister chromatid exchange and micronuclei in patients with type-1 diabetes mellitus. Mutat Res 2009. 676(1-2):1-4. [DOD] [CrossRef]
- Gelaleti RB, Damasceno DC, Lima PH, Salvadori DM, Calderon Ide M, Peracoli JC, Rudge MV. Oxidative DNA damage in diabetic and mild gestational hyperglycemic pregnant women. Diabetol Metab Syndr 2015. 7:1. [DOD] [CrossRef]
- Gelaleti RB, Damasceno DC, Salvadori DM, Marcondes JP, Lima PH, Morceli G, Calderon IM, Rudge MV. IRS-1 gene polymorphism and DNA damage in pregnant women with diabetes or mild gestational hyperglycemia. Diabetol Metab Syndr 2015. 7:30. [DOD] [CrossRef]
- Simone S, Gorin Y, Velagapudi C, Abboud HE, Habib SL. Mechanism of oxidative DNA damage in diabetes : tuberin inactivation and downregulation of DNA repair enzyme 8-oxo-7,8-dihydro-2'-deoxyguanosine-DNA glycosylase. Diabetes 2008. 57(10):2626-2636. [DOD] [CrossRef]
- Xu GW, Yao QH, Weng QF, Su BL, Zhang X, Xiong JH. Study of urinary 8-hydroxydeoxyguanosine as a biomarker of oxidative DNA damage in diabetic nephropathy patients. J Pharm Biomed Anal 2004. 36(1):101-104. [DOD] [CrossRef]
- Dong QY, Cui Y, Chen L, Song J, Sun L. Urinary 8-hydroxydeoxyguanosine levels in diabetic retinopathy patients. Eur J Ophthalmol 2008. 18(1):94-98. [DOD]
- Valavanidis A, Vlachogianni T, Fiotakis C. 8-hydroxy-2'-deoxyguanosine (8-OHdG): A critical biomarker of oxidative stress and carcinogenesis. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev 2009. 27(2):120-139. [DOD] [CrossRef]
- Orhan H, van Holland B, Krab B, Moeken J, Vermeulen NP, Hollander P, Meerman JH. Evaluation of a multi-parameter biomarker set for oxidative damage in man: increased urinary excretion of lipid, protein and DNA oxidation products after one hour of exercise. Free Radic Res 2004. 38(12):1269-1279. [DOD] [CrossRef]
- Lai CH, Liou SH, Lin HC, Shih TS, Tsai PJ, Chen JS, Yang T, Jaakkola JJ, Strickland PT. Exposure to traffic exhausts and oxidative DNA damage. Occup Environ Med 2005. 62(4):216-222. [DOD] [CrossRef]
- Bohr VA, Dianov GL. Oxidative DNA damage processing in nuclear and mitochondrial DNA. Biochimie 1999. 81(1-2):155-160. [DOD] [CrossRef]
- Yakes FM, Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci U S A 1997. 94(2):514-519. [DOD] [CrossRef]
- Gao Y, Katyal S, Lee Y, Zhao J, Rehg JE, Russell HR, McKinnon PJ. DNA ligase III is critical for mtDNA integrity but not Xrcc1-mediated nuclear DNA repair. Nature 2011. 471(7337):240-244. [DOD] [CrossRef]
- de Souza-Pinto NC, Wilson DM, Stevnsner TV, Bohr VA. Mitochondrial DNA, base excision repair and neurodegeneration. DNA Repair 2008. 7(7):1098-1109. [DOD] [CrossRef]
- Suematsu N, Tsutsui H, Wen J, Kang D, Ikeuchi M, Ide T, Hayashidani S, Shiomi T, Kubota T, Hamasaki N, Takeshita A. Oxidative stress mediates tumor necrosis factor-alpha-induced mitochondrial DNA damage and dysfunction in cardiac myocytes. Circulation 2003. 107(10):1418-1423. [DOD] [CrossRef]
- Madsen-Bouterse SA, Mohammad G, Kanwar M, Kowluru RA. Role of mitochondrial DNA damage in the development of diabetic retinopathy, and the metabolic memory phenomenon associated with its progression. Antioxid Redox Signal 2010. 13(6):797-805. [DOD] [CrossRef]
- Czajkaa AS, Gnudib L, Parsadea CK, Jonesa P, Reidc F, Malika AN. Altered mitochondrial function, mitochondrial DNA and reduced metabolic flexibility in patients with diabetic nephropathy. EBioMedicine 2015. 2(6):499-512. [DOD] [CrossRef]
- Merecz A, Markiewicz L, Sliwinska A, Kosmalski M, Kasznicki J, Drzewoski J, Majsterek I. Analysis of oxidative DNA damage and its repair in Polish patients with diabetes mellitus type 2: Role in pathogenesis of diabetic neuropathy. Adv Med Sci 2015. 60(2):220-230. [DOD] [CrossRef]
- Kasznicki J, Kosmalski M, Sliwinska A, Mrowicka M, Stanczyk M, Majsterek I, Drzewoski J. Evaluation of oxidative stress markers in pathogenesis of diabetic neuropathy. Mol Biol Rep 2012. 39(9):8669-8678. [DOD] [CrossRef]
- Lu J, Li X, Zhang M, Chen Z, Wang Y, Zeng C, Liu Z, Chen H. Regulation of MUTYH, a DNA repair enzyme, in renal proximal tubular epithelial cells. Oxid Med Cell Longev 2014. In press. [DOD]
- Cai Z, Chen H, Tao J, Guo W, Liu X, Zheng B, Sun W, Wang Y. Association of Base Excision Repair Gene Polymorphisms with ESRD Risk in a Chinese Population. Oxid Med Cell Longev 2012. 2012:10. [DOD]
- Cai Z, Guo W, Chen H, Tao J, Cao L, Sun W, Wang Y. Base excision repair gene polymorphisms are associated with inflammation in patients undergoing chronic hemodialysis. Biochem Biophys Res Commun 2012. 424(3):611-615. [DOD] [CrossRef]
- Bernstein C, Bernstein H, Payne CM, Garewal H. DNA repair/pro-apoptotic dual-role proteins in five major DNA repair pathways: fail-safe protection against carcinogenesis. Mutat Res 2002. 511(2):145-178. [DOD] [CrossRef]
- Ashkenazi A, Dixit VM. Apoptosis control by death and decoy receptors. Curr Opin Cell Biol 1999. 11(2):255-260. [DOD] [CrossRef]
- Eguchi Y, Shimizu S, Tsujimoto Y. Intracellular ATP Levels Determine Cell Death Fate by Apoptosis or Necrosis. Cancer Res 1997. 57(10):1835-1840. [DOD]
- Malhotra JD, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid Redox Signal 2007. 9(12):2277-2293. [DOD]
- Herman-Edelstein M, Thomas MC, Thallas-Bonke V, Saleem M, Cooper ME, Kantharidis P. Dedifferentiation of immortalized human podocytes in response to transforming growth Factor-beta: a model for diabetic podocytopathy. Diabetes 2011. 60(6):1779-1788. [DOD] [CrossRef]
- Sanchez-Nino MD, Sanz AB, Lorz C, Gnirke A, Rastaldi MP, Nair V, Egido J, Ruiz-Ortega M, Kretzler M, Ortiz A. BASP1 promotes apoptosis in diabetic nephropathy. J Am Soc Nephrol 2010. 21(4):610-621. [DOD] [CrossRef]
- Kumar D, Robertson S, Burns KD. Evidence of apoptosis in human diabetic kidney. Mol Cell Biochem 2004. 259(1-2):67-70. [DOD] [CrossRef]
- Kumar D, Zimpelmann J, Robertson S, Burns KD. Tubular and interstitial cell apoptosis in the streptozotocin-diabetic rat kidney. Nephron Exp Nephrol 2004. 96(3):E77-E88. [DOD] [CrossRef]
- Susztak K, Ciccone E, McCue P, Sharma K, Böttinger EP. Multiple metabolic hits converge on CD36 as novel mediator of tubular epithelial apoptosis in diabetic nephropathy. Plos Med 2005. 2(2):e45. [DOD] [CrossRef]
- Lorz C, Benito-Martín A, Boucherot A, Ucero AC, Rastaldi MP, Henger A, Armelloni S, Santamaría B, Berthier CC, Kretzler M, et al. The death ligand TRAIL in diabetic nephropathy. J Am Soc Nephrol 2008. 19(5):904-914. [DOD] [CrossRef]
- Sanchez-Nino MD, Benito-Martin A, Ortiz A. New paradigms in cell death in human diabetic nephropathy. Kidney Int 2010. 78(8):737-744. [DOD] [CrossRef]
- Chang YH, Lin KD, He SR, Hsieh MC, Hsiao JY, Shin SJ. Serum osteoprotegerin and tumor necrosis factor related apoptosis inducing-ligand (TRAIL) are elevated in type 2 diabetic patients with albuminuria and serum osteoprotegerin is independently associated with the severity of diabetic nephropathy. Metabolism 2011. 60(8):1064-1069. [DOD] [CrossRef]
- Verzola D, Gandolfo MT, Ferrario F, Rastaldi MP, Villaggio B, Gianiorio F, Giannoni M, Rimoldi L, Lauria F, Miji M, et al. Apoptosis in the kidneys of patients with type II diabetic nephropathy. Kidney Int 2007. 72(10):1262-1272. [DOD] [CrossRef]
- Baba K, Minatoguchi S, Sano H, Kagawa T, Murata I, Takemura G, Hirano T, Ohashi H, Takemura M, Fujiwara T, Fujiwara H. Involvement of apoptosis in patients with diabetic nephropathy: A study on plasma soluble Fas levels and pathological findings. Nephrology (Carlton) 2004. 9(2):94-99. [DOD] [CrossRef]
- Kelly DJ, Stein-Oakley A, Zhang Y, Wassef L, Maguire J, Koji T, Thomson N, Wilkinson-Berka JL, Gilbert RE. Fas-induced apoptosis is a feature of progressive diabetic nephropathy in transgenic (mRen-2)27 rats: attenuation with renin-angiotensin blockade. Nephrology (Carlton) 2004. 9(1):7-13. [DOD] [CrossRef]
- Justo P, Sanz AB, Lorz C, Egido J, Ortiz A. Lethal activity of FADD death domain in renal tubular epithelial cells. Kidney Int 2006. 69(12):2205-2211. [DOD] [CrossRef]
- Erkan E, De Leon M, Devarajan P. Albumin overload induces apoptosis in LLC-PK(1) cells. Am J Physiol Renal Physiol 2001. 280(6):F1107-F1114. [DOD]
- Cipollone F, Chiarelli F, Iezzi A, Fazia ML, Cuccurullo C, Pini B, De Cesare D, Torello M, Tumini S, Cuccurullo F, Mezzetti A. Relationship between reduced BCL-2 expression in circulating mononuclear cells and early nephropathy in type 1 diabetes. Int J Immunopathol Pharmacol 2005. 18(4):625-635. [DOD]
- Ortiz A, Ziyadeh FN, Neilson EG. Expression of apoptosis-regulatory genes in renal proximal tubular epithelial cells exposed to high ambient glucose and in diabetic kidneys. J Investig Medi 1997. 45(2):50-56. [DOD]
- Liu F, Brezniceanu ML, Wei CC, Chenier I, Sachetelli S, Zhang SL, Filep JG, Ingelfinger JR, Chan JS. Overexpression of angiotensinogen increases tubular apoptosis in diabetes. J Am Soc Nephrol 2008. 19(2):269-280. [DOD] [CrossRef]
- Qiu LQ, Sinniah R, I-Hong Hsu S. Downregulation of Bcl-2 by podocytes is associated with progressive glomerular injury and clinical indices of poor renal prognosis in human IgA nephropathy. J Am Soc Nephrol 2004. 15(1):79-90. [DOD] [CrossRef]
- Goumenos DS, Tsamandas AC, Kalliakmani P, Tsakas S, Sotsiou F, Bonikos DS, Vlachojannis JG. Expression of apoptosis-related proteins bcl-2 and bax along with transforming growth factor (TGF-beta1) in the kidney of patients with glomerulonephritides. Ren Fail 2004. 26(4):361-367. [DOD] [CrossRef]
- Lau GJ, Godin N, Maachi H, Lo CS, Wu SJ, Zhu JX, Brezniceanu ML, Chenier I, Fragasso-Marquis J, Lattouf JB, et al. Bcl-2-modifying factor induces renal proximal tubular cell apoptosis in diabetic mice. Diabetes 2012. 61(2):474-484. [DOD] [CrossRef]
- Gerecova G, Kopanicova J, Jaka P, Behalova L, Juhasova B, Bhatia-Kissova I, Forte M, Polcic P, Mentel M. BH3-only proteins Noxa, Bik, Bmf, and Bid activate Bax and Bak indirectly when studied in yeast model. FEMS Yeast Res 2013. 13(8):747-754. [DOD] [CrossRef]
- Rane MJ, Song Y, Jin S, Barati MT, Wu R, Kausar H, Tan Y, Wang Y, Zhou G, Klein JB, et al. Interplay between Akt and p38 MAPK pathways in the regulation of renal tubular cell apoptosis associated with diabetic nephropathy. Am J Physiol Renal Physiol 2010. 298(1):F49-F61. [DOD] [CrossRef]
- Wagener FA, Dekker D, Berden JH, Scharstuhl A, van der Vlag J. The role of reactive oxygen species in apoptosis of the diabetic kidney. Apoptosis 2009. 14(12):1451-1458. [DOD] [CrossRef]
- Song P, Yang S, Xiao L, Xu X, Tang C, Yang Y, Ma M, Zhu J, Liu F, Sun L. PKCdelta promotes high glucose induced renal tubular oxidative damage via regulating activation and translocation of p66Shc. Oxid Med Cell Longev 2014. 2014:746531. [DOD]
- Sun L, Xiao L, Nie J, Liu FY, Ling GH, Zhu XJ, Tang WB, Chen WC, Xia YC, Zhan M, et al. p66Shc mediates high-glucose and angiotensin II-induced oxidative stress renal tubular injury via mitochondrial-dependent apoptotic pathway. Am J Physiol Renal Physiol 2010. 299(5):F1014-F1025. [DOD] [CrossRef]
- Verzola D, Ratto E, Villaggio B, Parodi EL, Pontremoli R, Garibotto G, Viazzi F. Uric acid promotes apoptosis in human proximal tubule cells by oxidative stress and the activation of NADPH oxidase NOX 4. Plos One 2014. 9(12):e115210. [DOD] [CrossRef]
- Susnow N, Zeng L, Margineantu D, Hockenbery DM. Bcl-2 family proteins as regulators of oxidative stress. Semin Cancer Biol 2009. 19(1):42-49. [DOD] [CrossRef]
- D'Alessio M, De Nicola M, Coppola S, Gualandi G, Pugliese L, Cerella C, Cristofanon S, Civitareale P, Ciriolo MR, Bergamaschi A, et al. Oxidative Bax dimerization promotes its translocation to mitochondria independently of apoptosis. Faseb J 2005. 19(11):1504-1506. [DOD]
- Karch J, Molkentin JD. Regulated necrotic cell death: the passive aggressive side of Bax and Bak. Circ Res 2015. 116(11):1800-1809. [DOD] [CrossRef]
- Whelan RS, Konstantinidis K, Wei AC, Chen Y, Reyna DE, Jha S, Yang Y, Calvert JW, Lindsten T, Thompson CB, et al. Bax regulates primary necrosis through mitochondrial dynamics. Proc Natl Acad Sci U S A 2012. 109(17):6566-6571. [DOD] [CrossRef]
- Dewson G, Kluck RM. Mechanisms by which Bak and Bax permeabilise mitochondria during apoptosis. J Cell Sci 2009. 122(16):2801-2808. [DOD] [CrossRef]
- Lin SH, Perera MN, Nguyen T, Datskovskiy D, Miles M, Colombini M. Bax forms two types of channels, one of which is voltage-gated. Biophys J 2011. 101(9):2163-2169. [DOD] [CrossRef]
- Chen Y, Gui D, Chen J, He D, Luo Y, Wang N. Down-regulation of PERK-ATF4-CHOP pathway by Astragaloside IV is associated with the inhibition of endoplasmic reticulum stress-induced podocyte apoptosis in diabetic rats. Cell Physiol Biochem 2014. 33(6):1975-1987. [DOD] [CrossRef]
- Baban B, Liu JY, Mozaffari MS. Endoplasmic reticulum stress response and inflammatory cytokines in type 2 diabetic nephropathy: Role of indoleamine 2,3-dioxygenase and programmed death-1. Exp Mol Pathol 2013. 94(2):343-351. [DOD] [CrossRef]
- Brosius FC, Kaufman RJ. Is the ER stressed out in diabetic kidney disease? J Am Soc Nephrol 2008. 19(11):2040-2042. [DOD]
- Forbes JM, Coughlan MT, Cooper ME. Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes 2008. 57(6):1446-1454. [DOD] [CrossRef]
- Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 Regulates a Subset of Endoplasmic Reticulum Resident Chaperone Genes in the Unfolded Protein Response. Mol Cell Biol 2003. 23(21):7448-7459. [DOD] [CrossRef]
- Chen Y, Brandizzi F. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol 2013. 23(11):547-555. [DOD] [CrossRef]
- Fonseca SG, Gromada J, Urano F. Endoplasmic reticulum stress and pancreatic beta-cell death. Trends Endocrinol Metab 2011. 22(7):266-274. [DOD]
- Laybutt DR, Preston AM, Akerfeldt MC, Kench JG, Busch AK, Biankin AV, Biden TJ. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia 2007. 50(4):752-763. [DOD] [CrossRef]
- Lindenmeyer MT, Rastaldi MP, Ikehata M, Neusser MA, Kretzler M, Cohen CD, Schlöndorff D. Proteinuria and hyperglycemia induce endoplasmic reticulum stress. J Am Soc Nephrol 2008. 19(11):2225-2236. [DOD] [CrossRef]
- Zhuang A, Forbes JM. Stress in the kidney is the road to pERdition: is endoplasmic reticulum stress a pathogenic mediator of diabetic nephropathy? J Endocrinol 2014. 222(3):R97-R111. [DOD]
- Inagi R. Endoplasmic reticulum stress as a progression factor for kidney injury. Curr Opin Pharmacol 2010. 10(2):156-165. [DOD] [CrossRef]
- Brezniceanu ML, Lau CJ, Godin N, Chenier I, Duclos A, Ethier J, Filep JG, Ingelfinger JR, Zhang SL, Chan JS. Reactive oxygen species promote caspase-12 expression and tubular apoptosis in diabetic nephropathy. J Am Soc Nephrol 2010. 21(6):943-954. [DOD] [CrossRef]
- Cheema MU, Damkier HH, Nielsen J, Poulsen ET, Enghild JJ, Fenton RA, Praetorius J. Distal renal tubules are deficient in aggresome formation and autophagy upon aldosterone administration. Plos One 2014. 9(7):e101258. [DOD] [CrossRef]
- Feissner RF, Skalska J, Gaum WE, Sheu SS. Crosstalk signaling between mitochondrial Ca(2+) and ROS. Front Biosci (Landmark Ed) 2009. 14:1197-1218. [DOD] [CrossRef]
- Grimm S. The ER–mitochondria interface: The social network of cell death. Biochim Biophys Acta 2012. 1823(2):327-334. [DOD] [CrossRef]
- Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol 2004. 287(4):C817-C833. [DOD] [CrossRef]
- Pospisilik JA, Knauf C, Joza N, Benit P, Orthofer M, Cani PD, Ebersberger I, Nakashima T, Sarao R, Neely G, et al. Targeted deletion of AIF decreases mitochondrial oxidative phosphorylation and protects from obesity and diabetes. Cell 2007. 131(3):476-491. [DOD] [CrossRef]
- Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta 2013. 1833(12):3460-3470. [DOD] [CrossRef]
- Wang X, Olberding KE, White C, Li C. Bcl-2 proteins regulate ER membrane permeability to luminal proteins during ER stress-induced apoptosis. Cell Death Differ 2011. 18(1):38-47. [DOD] [CrossRef]
- Zong WX, Li C, Hatzivassiliou G, Lindsten T, Yu QC, Yuan J, Thompson CB. Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis. J Cell Biol 2003. 162(1):59-69. [DOD] [CrossRef]
- Kennedy D, Mnich K, Samali A. Heat shock preconditioning protects against ER stress-induced apoptosis through the regulation of the BH3-only protein BIM. FEBS Open Bio 2014. 4:813-821. [DOD] [CrossRef]
- Morishima N, Nakanishi K, Tsuchiya K, Shibata T, Seiwa E. Translocation of Bim to the endoplasmic reticulum (ER) mediates ER stress signaling for activation of caspase-12 during ER stress-induced apoptosis. J Biol Chem 2004. 279(48):50375-50381. [DOD] [CrossRef]
- Lin ML, Chen SS, Huang RY, Lu YC, Liao YR, Reddy MV, Lee CC, Wu TS. Suppression of PI3K/Akt signaling by synthetic bichalcone analog TSWU-CD4 induces ER stress- and Bax/Bak-mediated apoptosis of cancer cells. Apoptosis 2014. 19(11):1637-1653. [DOD] [CrossRef]
- Matsuzaki S, Hiratsuka T, Kuwahara R, Katayama T, Tohyama M. Caspase-4 is partially cleaved by calpain via the impairment of Ca2+ homeostasis under the ER stress. Neurochem Int 2010. 56(2):352-356. [DOD] [CrossRef]
- Sansanwal P, Kambham N, Sarwal MM. Caspase-4 may play a role in loss of proximal tubules and renal injury in nephropathic cystinosis. Pediatr Nephrol 2010. 25(1):105-109. [DOD] [CrossRef]
- Sanchez-Nino MD, Sanz AB, Ortiz A. Caspase-12 and Diabetic Nephropathy: From Mice to Men? J Am Soc Nephrol 2010. 21(6):886-888. [DOD]
- Deegan S, Saveljeva S, Logue SE, Pakos-Zebrucka K, Gupta S, Vandenabeele P, Bertrand MJ, Samali A. Deficiency in the mitochondrial apoptotic pathway reveals the toxic potential of autophagy under ER stress conditions. Autophagy 2014. 10(11):1921-1936. [DOD] [CrossRef]
- Ueda M, Li S, Itoh M, Hayakawa-Yano Y, Wang MX, Hayakawa M, Hasebe-Matsubara R, Ohta K, Ohta E, Mizuno A, et al. Polyglutamine expansion disturbs the endoplasmic reticulum formation, leading to caspase-7 activation through Bax. Biochem Biophys Res Commun 2014. 443(4):1232-1238. [DOD] [CrossRef]
- Gill MB, Bockhorst K, Narayana P, Perez-Polo JR. Bax shuttling after neonatal hypoxia-ischemia: hyperoxia effects. J Neurosci Res 2008. 86(16):3584-3604. [DOD] [CrossRef]
- Bonneau B, Prudent J, Popgeorgiev N, Gillet G. Non-apoptotic roles of Bcl-2 family: the calcium connection. Biochim Biophys Acta 2013. 1833(7):1755-1765. [DOD] [CrossRef]
- Small DM, Bennett NC, Roy S, Gabrielli BG, Johnson DW, Gobe GC. Oxidative stress and cell senescence combine to cause maximal renal tubular epithelial cell dysfunction and loss in an in vitro model of kidney disease. Nephron Exp Nephrol 2012. 122(3-4):123-130. [DOD] [CrossRef]
- Frippiat C, Chen QM, Zdanov S, Magalhaes JP, Remacle J, Toussaint O. Subcytotoxic H2O2 stress triggers a release of transforming growth factor-beta1, which induces biomarkers of cellular senescence of human diploid fibroblasts. J Biol Chem 2001. 276(4):2531-2537. [DOD] [CrossRef]
- de Haan JB, Bladier C, Lotfi-Miri M, Taylor J, Hutchinson P, Crack PJ, Hertzog P, Kola I. Fibroblasts derived from Gpx1 knockout mice display senescent-like features and are susceptible to H2O2-mediated cell death. Free Radic Biol Med 2004. 36(1):53-64. [DOD] [CrossRef]
- Zhu K, Kakehi T, Matsumoto M, Iwata K, Ibi M, Ohshima Y, Zhang J, Liu J, Wen X, Taye A, et al. NADPH oxidase NOX1 is involved in activation of protein kinase C and premature senescence in early stage diabetic kidney. Free Radic Biol Med 2015. 83:21-30. [DOD] [CrossRef]
- Verzola D, Gandolfo MT, Gaetani G, Ferraris A, Mangerini R, Ferrario F, Villaggio B, Gianiorio F, Tosetti F, Weiss U, et al. Accelerated senescence in the kidneys of patients with type 2 diabetic nephropathy. Am J Physiol Renal Physiol 2008. 295(5):F1563-F1573. [DOD] [CrossRef]
- Lee HC, Yin PH, Lu CY, Chi CW, Wei YH. Increase of mitochondria and mitochondrial DNA in response to oxidative stress in human cells. Biochem J 2000. 348(Pt 2):425-432. [DOD]
- Malik AN, Shahni R, Iqbal MM. Increased peripheral blood mitochondrial DNA in type 2 diabetic patients with nephropathy. Diabetes Res Clin Pract 2009. 86(2):E22-E24. [DOD] [CrossRef]
- Nakamura T, Ushiyama C, Suzuki S, Hara M, Shimada N, Ebihara I, Koide H. Urinary excretion of podocytes in patients with diabetic nephropathy. Nephrol Dial Transplant 2000. 15(9):1379-1383. [DOD] [CrossRef]
- Soltoff SP. ATP and the regulation of renal cell function. Annu Rev Physiol 1986. 48:9-31. [DOD] [CrossRef]
- Halestrap AP. What is the mitochondrial permeability transition pore? J Mol Cell Cardiol 2009. 46(6):821-831. [DOD]
- Halestrap AP, Pasdois P. The role of the mitochondrial permeability transition pore in heart disease. Biochim Biophys Acta 2009. 1787(11):1402-1415. [DOD] [CrossRef]
- Bernardi P. The mitochondrial permeability transition pore: a mystery solved? Front Physiol 2013. 4:95. [DOD]
- Karch J, Molkentin JD. Identifying the components of the elusive mitochondrial permeability transition pore. Proc Natl Acad Sci U S A 2014. 111(29):10396-10397. [DOD] [CrossRef]
- Takebayashi S, Kaneda K. Mitochondrial derangement: possible initiator of microalbuminuria in NIDDM. J Diabet Complications 1991. 5(2-3):104-106. [DOD] [CrossRef]
- Kaneda K, Iwao J, Sakata N, Takebayashi S. Correlation between mitochondrial enlargement in renal proximal tubules and microalbuminuria in rats with early streptozotocin-induced diabetes. Acta Pathol Jpn 1992. 42(12):855-860. [DOD]
- Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci U S A 2006. 103(8):2653-2658. [DOD] [CrossRef]
- Zoratti M, Szabo I. The mitochondrial permeability transition. Biochim Biophys Acta 1995. 1241(2):139-176. [DOD] [CrossRef]
- Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J 1999. 341(Pt 2):233-249. [DOD] [CrossRef]
- Halestrap AP. Calcium, mitochondria and reperfusion injury: a pore way to die. Biochem Soc Trans 2006. 34(Pt 2):232-237. [DOD] [CrossRef]
- Armstrong JS. The role of the mitochondrial permeability transition in cell death. Mitochondrion 2006. 6(5):225-234. [DOD] [CrossRef]
- Zorov DB, Juhaszova M, Yaniv Y, Nuss HB, Wang S, Sollott SJ. Regulation and pharmacology of the mitochondrial permeability transition pore. Cardiovasc Res 2009. 83(2):213-225. [DOD] [CrossRef]
- Baines CP. The molecular composition of the mitochondrial permeability transition pore. J Mol Cell Cardiol 2009. 46(6):850-857. [DOD] [CrossRef]
- Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol 2007. 9(5):550-555. [DOD] [CrossRef]
- Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 2004. 427(6973):461-465. [DOD] [CrossRef]
- Krauskopf A, Eriksson O, Craigen WJ, Forte MA, Bernardi P. Properties of the permeability transition in VDAC1(-/-) mitochondria. Biochim Biophys Acta 2006. 1757(5-6):590-595. [DOD] [CrossRef]
- Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci U S A 2005. 102(34):12005-12010. [DOD] [CrossRef]
- Woodfield K, Ruck A, Brdiczka D, Halestrap AP. Direct demonstration of a specific interaction between cyclophilin-D and the adenine nucleotide translocase confirms their role in the mitochondrial permeability transition. Biochem J 1998. 336(Pt 2):287-290. [DOD] [CrossRef]
- Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005. 434(7033):658-662. [DOD] [CrossRef]
- Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 2005. 434(7033):652-658. [DOD] [CrossRef]
- Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, Glick GD, Petronilli V, Zoratti M, Szabo I, et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci U S A 2013. 110(15):5887-5892. [DOD] [CrossRef]
- Alavian KN, Beutner G, Lazrove E, Sacchetti S, Park HA, Licznerski P, Li H, Nabili P, Hockensmith K, Graham M, et al. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc Natl Acad Sci U S A 2014. 111(29):10580-10585. [DOD] [CrossRef]
- Takeyama N, Miki S, Hirakawa A, Tanaka T. Role of the mitochondrial permeability transition and cytochrome C release in hydrogen peroxide-induced apoptosis. Exp Cell Res 2002. 274(1):16-24. [DOD] [CrossRef]
- Batandier C, Leverve X, Fontaine E. Opening of the mitochondrial permeability transition pore induces reactive oxygen species production at the level of the respiratory chain complex I. J Biol Chem 2004. 279(17):17197-17204. [DOD] [CrossRef]
- Scorrano L, Ashiya M, Buttle K, Weiler S, Oakes SA, Mannella CA, Korsmeyer SJ. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev Cell 2002. 2(1):55-67. [DOD] [CrossRef]
- Boveris A, Oshino N, Chance B. The cellular production of hydrogen peroxide. Biochem J 1972. 128(3):617-630. [DOD] [CrossRef]
- Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol 2003. 552(Pt 2):335-344. [DOD] [CrossRef]
- Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial ROS-induced ROS release: an update and review. Biochim Biophys Acta 2006. 1757(5-6):509-517. [DOD] [CrossRef]
- Whelan RS, Konstantinidis K, Wei AC, Chen Y, Reyna DE, Jha S, Yang Y, Calvert JW, Lindsten T, Thompson CB, et al. Bax regulates primary necrosis through mitochondrial dynamics. Proc Natl Acad Sci U S A 2012. 109(17):6566-6571. [DOD] [CrossRef]
- Devalaraja-Narashimha K, Diener AM, Padanilam BJ. Cyclophilin D gene ablation protects mice from ischemic renal injury. Am J Physiol Renal Physiol 2009. 297(3):F749-F759. [DOD] [CrossRef]
- Gooch JL, Barnes JL, Garcia S, Abboud HE. Calcineurin is activated in diabetes and is required for glomerular hypertrophy and ECM accumulation. Am J Physiol Renal Physiol 2003. 284(1):F144-F154. [DOD] [CrossRef]
- Oliveira PJ, Esteves TC, Seica R, Moreno AJ, Santos MS. Calcium-dependent mitochondrial permeability transition is augmented in the kidney of Goto-Kakizaki diabetic rat. Diabetes Metab Res Rev 2004. 20(2):131-136. [DOD] [CrossRef]
- Smith RA, Porteous CM, Gane AM, Murphy MP. Delivery of bioactive molecules to mitochondria in vivo. Proc Natl Acad Sci U S A 2003. 100(9):5407-5412. [DOD] [CrossRef]
- Mukhopadhyay P, Horvath B, Zsengeller Z, Zielonka J, Tanchian G, Holovac E, Kechrid M, Patel V, Stillman IE, Parikh SM, et al. Mitochondrial-targeted antioxidants represent a promising approach for prevention of cisplatin-induced nephropathy. Free Radic Biol Med 2012. 52(2):497-506. [DOD] [CrossRef]
- Tabara LC, Poveda J, Martin-Cleary C, Selgas R, Ortiz A, Sanchez-Nino MD. Mitochondria-targeted therapies for acute kidney injury. Expert Rev Mol Med 2014. 16:e13. [DOD] [CrossRef]
- Subramanian S, Kalyanaraman B, Migrino RQ. Mitochondrially targeted antioxidants for the treatment of cardiovascular diseases. Recent Pat Cardiovasc Drug Discov 2010. 5(1):54-65. [DOD] [CrossRef]
- Chacko BK, Reily C, Srivastava A, Johnson MS, Ye Y, Ulasova E, Agarwal A, Zinn KR, Murphy MP, Kalyanaraman B, Darley-Usmar V. Prevention of diabetic nephropathy in Ins2(+/)(-)(AkitaJ) mice by the mitochondria-targeted therapy MitoQ. Biochem J 2010. 432(1):9-19. [DOD] [CrossRef]
- Bolisetty S, Traylor A, Zarjou A, Johnson MS, Benavides GA, Ricart K, Boddu R, Moore RD, Landar A, Barnes S, et al. Mitochondria-targeted heme oxygenase-1 decreases oxidative stress in renal epithelial cells. Am J Physiol Renal Physiol 2013. 305(3):F255-F264. [DOD] [CrossRef]
- Nakamura Y, Feng Q, Kumagai T, Torikai K, Ohigashi H, Osawa T, Noguchi N, Niki E, Uchida K. Ebselen, a glutathione peroxidase mimetic seleno-organic compound, as a multifunctional antioxidant. Implication for inflammation-associated carcinogenesis. J Biol Chem 2002. 277(4):2687-2694. [DOD] [CrossRef]
- Tan SM, Sharma A, Yuen DY, Stefanovic N, Krippner G, Mugesh G, Chai Z, de Haan JB. The modified selenenyl amide, M-hydroxy ebselen, attenuates diabetic nephropathy and diabetes-associated atherosclerosis in ApoE/GPx1 double knockout mice. Plos One 2013. 8(7):e69193. [DOD] [CrossRef]
- Filipovska A, Kelso GF, Brown SE, Beer SM, Smith RA, Murphy MP. Synthesis and characterization of a triphenylphosphonium-conjugated peroxidase mimetic. Insights into the interaction of ebselen with mitochondria. J Biol Chem 2005. 280(25):24113-24126. [DOD] [CrossRef]
- Szeto HH, Liu S, Soong Y, Wu D, Darrah SF, Cheng FY, Zhao Z, Ganger M, Tow CY, Seshan SV. Mitochondria-targeted peptide accelerates ATP recovery and reduces ischemic kidney injury. J Am Soc Nephrol 2011. 22(6):1041-1052. [DOD] [CrossRef]
- Chakrabarti AK, Feeney K, Abueg C, Brown DA, Czyz E, Tendera M, Janosi A, Giugliano RP, Kloner RA, Weaver WD, et al. Rationale and design of the EMBRACE STEMI study: a phase 2a, randomized, double-blind, placebo-controlled trial to evaluate the safety, tolerability and efficacy of intravenous Bendavia on reperfusion injury in patients treated with standard therapy including primary percutaneous coronary intervention and stenting for ST-segment elevation myocardial infarction. Am Heart J 2013. 165(4):509-514. [DOD] [CrossRef]
- Javadov S, Karmazyn M, Escobales N. Mitochondrial permeability transition pore opening as a promising therapeutic target in cardiac diseases. J Pharmacol Exp Ther 2009. 330(3):670-678. [DOD] [CrossRef]
- Halestrap AP, Davidson AM. Inhibition of Ca2(+)-induced large-amplitude swelling of liver and heart mitochondria by cyclosporin is probably caused by the inhibitor binding to mitochondrial-matrix peptidyl-prolyl cis-trans isomerase and preventing it interacting with the adenine nucleotide translocase. Biochem J 1990. 268(1):153-160. [DOD] [CrossRef]
- Tanveer A, Virji S, Andreeva L, Totty NF, Hsuan JJ, Ward JM, Crompton M. Involvement of cyclophilin D in the activation of a mitochondrial pore by Ca2+ and oxidant stress. Eur J Biochem 1996. 238(1):166-172. [DOD] [CrossRef]
- Piot C, Croisille P, Staat P, Thibault H, Rioufol G, Mewton N, Elbelghiti R, Cung TT, Bonnefoy E, Angoulvant D, et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med 2008. 359(5):473-481. [DOD] [CrossRef]
- Li C, Lim SW, Sun BK, Yang CW. Chronic cyclosporine nephrotoxicity: new insights and preventive strategies. Yonsei Med J 2004. 45(6):1004-1016. [DOD] [CrossRef]
- Ptak RG, Gallay PA, Jochmans D, Halestrap AP, Ruegg UT, Pallansch LA, Bobardt MD, de Bethune MP, Neyts J, De Clercq E, et al. Inhibition of human immunodeficiency virus type 1 replication in human cells by Debio-025, a novel cyclophilin binding agent. Antimicrob Agents Chemother 2008. 52(4):1302-1317. [DOD] [CrossRef]
- Tiepolo T, Angelin A, Palma E, Sabatelli P, Merlini L, Nicolosi L, Finetti F, Braghetta P, Vuagniaux G, Dumont JM, et al. The cyclophilin inhibitor Debio 025 normalizes mitochondrial function, muscle apoptosis and ultrastructural defects in Col6a1(-/-) myopathic mice. Br J Pharmacol 2009. 157(6):1045-1052. [DOD] [CrossRef]
- Angelin A, Tiepolo T, Sabatelli P, Grumati P, Bergamin N, Golfieri C, Mattioli E, Gualandi F, Ferlini A, Merlini L, et al. Mitochondrial dysfunction in the pathogenesis of Ullrich congenital muscular dystrophy and prospective therapy with cyclosporins. Proc Natl Acad Sci U S A 2007. 104(3):991-996. [DOD] [CrossRef]
- Tiepolo T, Angelin A, Palma E, Sabatelli P, Merlini L, Nicolosi L, Finetti F, Braghetta P, Vuagniaux G, Dumont JM, et al. The cyclophilin inhibitor Debio 025 normalizes mitochondrial function, muscle apoptosis and ultrastructural defects in Col6a1-/- myopathic mice. Br J Pharmacol 2009. 157(6):1045-1052. [DOD] [CrossRef]
- Quarato G, D'Aprile A, Gavillet B, Vuagniaux G, Moradpour D, Capitanio N, Piccoli C. The cyclophilin inhibitor alisporivir prevents hepatitis C virus-mediated mitochondrial dysfunction. Hepatology 2012. 55(5):1333-1343. [DOD] [CrossRef]
- Liang HL, Sedlic F, Bosnjak Z, Nilakantan V. SOD1 and MitoTEMPO partially prevent mitochondrial permeability transition pore opening, necrosis, and mitochondrial apoptosis after ATP depletion recovery. Free Radic Biol Med 2010. 49(10):1550-1560. [DOD] [CrossRef]
- Wang Z, Bao H, Ge Y, Zhuang S, Peng A, Gong R. Pharmacological targeting of GSK3beta confers protection against podocytopathy and proteinuria by desensitizing mitochondrial permeability transition. Br J Pharmacol 2015. 172(3):895-909. [DOD] [CrossRef]
- de Zeeuw D, Audhya P, Bakris GL, Chin M, Christ-Schmidt H, Goldsberry A, Houser M, Krauth M, Lambers Heerspink HJ, McMurray JJ, et al. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N Engl J Med 2013. 369(26):2492-2503. [DOD] [CrossRef]
- Tan SM, Sharma A, Stefanovic N, Yuen DY, Karagiannis TC, Meyer C, Ward KW, Cooper ME, de Haan JB. Derivative of bardoxolone methyl, dh404, in an inverse dose-dependent manner lessens diabetes-associated atherosclerosis and improves diabetic kidney disease. Diabetes 2014. 63(9):3091-3103. [DOD] [CrossRef]
- Trujillo J, Chirino YI, Molina-Jijon E, Anderica-Romero AC, Tapia E, Pedraza-Chaverri J. Renoprotective effect of the antioxidant curcumin: Recent findings. Redox Biol 2013. 1:448-456. [DOD] [CrossRef]
- Jiang T, Huang Z, Lin Y, Zhang Z, Fang D, Zhang DD. The protective role of Nrf2 in streptozotocin-induced diabetic nephropathy. Diabetes 2010. 59(4):850-860. [DOD] [CrossRef]
- Xu Y, Nie L, Yin YG, Tang JL, Zhou JY, Li DD, Zhou SW. Resveratrol protects against hyperglycemia-induced oxidative damage to mitochondria by activating SIRT1 in rat mesangial cells. Toxicol Appl Pharmacol 2012. 259(3):395-401. [DOD] [CrossRef]
- Alhaider AA, Korashy HM, Sayed-Ahmed MM, Mobark M, Kfoury H, Mansour MA. Metformin attenuates streptozotocin-induced diabetic nephropathy in rats through modulation of oxidative stress genes expression. Chem Biol Interact 2011. 192(3):233-242. [DOD] [CrossRef]
- Pal PB, Sinha K, Sil PC. Mangiferin attenuates diabetic nephropathy by inhibiting oxidative stress mediated signaling cascade, TNFalpha related and mitochondrial dependent apoptotic pathways in streptozotocin-induced diabetic rats. Plos One 2014. 9(9):e107220. [DOD] [CrossRef]
- Sedeek M, Gutsol A, Montezano AC, Burger D, Nguyen Dinh Cat A, Kennedy CR, Burns KD, Cooper ME, Jandeleit-Dahm K, Page P, et al. Renoprotective effects of a novel Nox1/4 inhibitor in a mouse model of Type 2 diabetes. Clin Sci (Lond) 2013. 124(3):191-202. [DOD] [CrossRef]
- ten Freyhaus H, Huntgeburth M, Wingler K, Schnitker J, Baumer AT, Vantler M, Bekhite MM, Wartenberg M, Sauer H, Rosenkranz S. Novel Nox inhibitor VAS2870 attenuates PDGF-dependent smooth muscle cell chemotaxis, but not proliferation. Cardiovasc Res 2006. 71(2):331-341. [DOD] [CrossRef]
- Lv M, Chen Z, Hu G, Li Q. Therapeutic strategies of diabetic nephropathy: recent progress and future perspectives. Drug Discov Today 2015. 20(3):332-346. [DOD] [CrossRef]
This article has been cited by other articles:

|
Potential Biochemical Mechanisms of Lung Injury in Diabetes
Zheng H, Wu J, Jin Z, Yan LJ
Aging Dis 2017. 8(1):7-16
|
|
|
Experimental study on Tangshenjiangzhuo granules treatment of rats
with early diabetic nephropathy
Xu XW, Xu XX
J Hainen Med Univ 2017. 23(3):13-16
|
|

|
HRD1-Mediated IGF-1R Ubiquitination Contributes to Renal Protection of Resveratrol in db/db Mice
Yan C, Xu W, Huang Y, Li M, Shen Y, You H, Liang X
Mol Endocrinol 2016. 30(6):600-613
|
|
|
Measuring redox-active species in cells and tissues
Witting PK
Am J Physiol Regul Integr Comp Physiol 2016. 310(7):R547-R548
|
|

|
Effect of papaya supplementation on oxidative stress markers in Parkinson's disease
Bolner A, Micciolo R, Bosello O, Nordera G
Oxid Antioxid Med Sci 2016. 5(2):49-55
|
|
|


)
)

