Review
| Rev Diabet Stud,
2009,
6(1):13-36 |
DOI 10.1900/RDS.2009.6.13 |
Adenosine Monophosphate-Activated Protein Kinase (AMPK) as a New Target for Antidiabetic Drugs: A Review on Metabolic, Pharmacological and Chemical Considerations
Arie Gruzman, Gali Babai, Shlomo Sasson
Department of Pharmacology, School of Pharmacy, Faculty of Medicine, The Hebrew University, Jerusalem 91120, Israel
Address correspondence to: Shlomo Sasson, e-mail: ShlomoSasson@huji.ac.il
Manuscript submitted April 10, 2009; resubmitted May 7, 2009; accepted May 9, 2009.
Keywords: diabetes, hyperglycemia, AMP, antihyperglycemic, drug target, energy metabolism, glucose transport, skeletal muscle, D-xylose
Abstract
In view of the epidemic nature of type 2 diabetes and the substantial rate of failure of current oral antidiabetic drugs the quest for new therapeutics is intensive. The adenosine monophosphate-activated protein kinase (AMPK) is an important regulatory protein for cellular energy balance and is considered a master switch of glucose and lipid metabolism in various organs, especially in skeletal muscle and liver. In skeletal muscles, AMPK stimulates glucose transport and fatty acid oxidation. In the liver, it augments fatty acid oxidation and decreases glucose output, cholesterol and triglyceride synthesis. These metabolic effects induced by AMPK are associated with lowering blood glucose levels in hyperglycemic individuals. Two classes of oral antihyperglycemic drugs (biguanidines and thiazolidinediones) have been shown to exert some of their therapeutic effects by directly or indirectly activating AMPK. However, side effects and an acquired resistance to these drugs emphasize the need for the development of novel and efficacious AMPK activators. We have recently discovered a new class of hydrophobic D-xylose derivatives that activates AMPK in skeletal muscles in a non insulin-dependent manner. One of these derivatives (2,4;3,5-dibenzylidene-D-xylose-diethyl-dithioacetal) stimulates the rate of hexose transport in skeletal muscle cells by increasing the abundance of glucose transporter-4 (GLUT-4) in the plasma membrane through activation of AMPK. This compound reduces blood glucose levels in diabetic mice and therefore offers a novel strategy of therapeutic intervention strategy in type 2 diabetes. The present review describes various classes of chemically-related compounds that activate AMPK by direct or indirect interactions and discusses their potential for candidate antihyperglycemic drug development.
The structure of the AMPK complex
The enzyme AMP-activated protein kinase (AMPK) is expressed in all eukaryotic cells as a heterotrimeric complex. The heteromeric structure and functions of this enzyme complex have been widely investigated. In this section, we focus on structural elements in this complex relevant for its interaction with synthetic pharmaceuticals.
The AMPK complex combines two regulatory subunits (β, 30 kDa; γ, 38-63 kDa) and a catalytic subunit (α, 63 kDa). These subunits are encoded by different genes and several isoforms of each have been discovered: α1, α2, β1, β2, γ1, γ2 and γ3 [1]. Theoretically, there are 12 possible heterotrimeric combinations of AMPK. Moreover, variable gene splicing of these gene products also contributes to the diversity of the AMPK complex composition. It appears that different types of cells and tissues express distinct combinations of the complex. For instance, human skeletal muscles display three complexes in which the β2 subunit is conserved: α2β2γ1, α1β2γ1 and α2β2γ3 [2]. Among these three complexes, the latter is predominantly associated with exercise-induced AMPK activation in human skeletal muscles, while the function of the other is less defined [3]. The β1 subunit is more ubiquitously expressed and is found in other tissues such as the liver [4].
The first crystal structure of a heterotrimeric AMPK complex was obtained from Saccharomyces cerevisae [5]. However, a full length crystal structure of mammalian AMPK has not yet been obtained. Therefore, structural analyses of the human AMPK subunits are largely based on partial domain structure identification and analysis of interactions among proteolytic truncated fragments of these subunits [6]. Detailed structural analysis of these complexes will enhance the effort to design, synthesize and test novel molecules that interact with distinct AMPK complexes expressed in different cells and organs.
The α subunit of the AMPK complex
This catalytic subunit contains a classical serine/threonine protein kinase domain close to the N-terminal [7]. Free catalytic α subunits are usually inactive due to the presence of an autoinhibitory domain that is located in the center of this subunit. When the α subunit forms a functional complex with the β and γ subunits, this autoinhibitory function no longer blocks the catalytic function. Interestingly, Pang et al. have recently suggested that small molecules can directly relieve this inherent autoinhibition in the α subunit, rendering the complex constitutively active and possibly resistant to intracellular degradation [8]. Although α1 and α2 subunits share similar substrate specificity [9], α2 is typically localized in the nucleus whereas α1 is predominantly found in the cytoplasm [10]. It has been shown in HeLa cells that certain stressful conditions induce α1 isoform translocation to the nucleus [11]. This compartmentalization and subcellular re-distribution may explain some diverse functions attributed to AMPK in cells.
The β subunit of the AMPK complex
The C-terminus of the β subunit contains recognition sequences that interact with the two other AMPK subunits and serves as the scaffold of the heterotrimeric complex. In addition, the central part of the β subunit contains a specific sequence that binds to glycogen particles. It has been proposed that this interaction is closely associated with a tight regulation of glycogen metabolism [12]. A myristoylation site located in the N-terminus of this subunit functions as a molecular switch for reversible membrane binding. The functional significance of this membrane docking has been demonstrated when a point mutation in the myristoylation site resulted in a 4-fold increase in the activity of the AMPK complex [13]. A direct autoinhibitory domain located in the N-terminal part of the β subunit has a critical role in autoregulating the activity of the complex [14]. In addition, the nuclear localization of the AMPK complex has been associated with targeted serine-phosphorylations in the β subunit [13]. These recent findings indicate that in addition to its established scaffold function, the β subunit also has important autoregulatory functions in the AMPK complex.
The γ subunit of the AMPK complex
Four tandem repeats of cystathionine-β-synthase sequences (CBS) that are located in the N-terminus of this subunit form two Bateman domains, which selectively bind adenosine containing molecules, such as AMP or ATP [15, 16]. Upon binding, the phosphate group of AMP/ATP lies in a groove on the surface of the γ subunit and interacts with basic residues of amino acids there [6]. Mutations in these basic amino acids result in several human hereditary metabolic disorders, such as impaired cardiac glycogen storage [17]. The binding of AMP to these Bateman domains activates AMPK, whereas ATP binding antagonizes this process. The two Bateman domains bind two pairs of AMP molecules in a positive cooperative manner, suggesting that the second domain is inaccessible for binding unless two AMP molecules occupy the CBS binding sites in the first Bateman domain. This allosteric interaction increases the sensitivity of the AMPK complex to minute changes in intracellular levels of AMP. In contrast, the binding of ATP molecules to these domains antagonizes activation of the complex. These disparate allosteric modulations in the complex, induced by the binding AMP or ATP, form the molecular basis for the metabolic switch function of AMPK and a plausible target for synthetic activators [16].
Mutations in the hydrophobic patch in the γ subunit (Arg171-Phe179), which interacts with the glycogen binding loop of the β subunits, substantially impede AMP-dependent activation of the AMPK complex [14]. It seems, therefore, that the activation process of the complex is also influenced by the γ subunit. Of interest is the finding that the γ2 subunit confers the complex with the greatest AMP dependency in comparison with the γ3 subunit, which moderately affects the sensitivity to AMP [18].
Activation of the AMPK complex
Among stressful conditions, which deplete ATP stores and increase AMP content in cells, heat shock, hypoxia, hyperosmolarity, glucose deprivation, ischemia and prolonged contraction of skeletal muscles have been studied most intensively [19, 20]. It is, however, important to note that some conditions known to activate AMPK in cultured cells are physiologically extreme. For instance, exaggerated hyperosmolarity or severe glucose deprivation are not compatible with real life situations. AMPK is activated in an AMP-dependent manner by targeted phosphorylation of Thr172 in the α subunit [21, 22]. The binding of AMP to the CBS domain in the γ subunit induces conformational changes in the hetrotrimeric complex that promote the phosphorylation of this Thr172 moiety by several upstream kinases. The dissociation of the AMP molecules is followed by the binding of ATP, which then increases the susceptibility of pThr172 to dephosphorylation by protein phosphatases [16].
The "energy charge hypothesis" assigns AMP regulatory metabolic functions in eukaryotic cells [23]. AMP is a very sensitive metabolic indicator of cellular energy metabolism due to the function of adenylate kinases in cells. These enzymes interconvert two molecules of ADP to AMP and ATP [21]. Because these kinases function at near equilibrium, the AMP:ATP ratio in cells varies as the square of the ADP:ATP ratio. When ATP is consumed or depleted and not adequately replenished, the ratio of ADP:ATP rises and drives the adenylate kinase reaction forward to generate ATP and AMP. The latter then further activates the AMPK pathway to conserve energy stores in cells. When cellular ATP levels are high enough and the generation of AMP is reduced, the former molecule preferentially binds to AMPK and blocks it [23]. Because the affinity of ATP binding to the Bateman domain in the γ subunit is lower than that of AMP, relatively higher concentrations of the former are required to attenuate the function of the complex [24]. The recent finding that only a small fraction of the total amount of cellular AMPK is amenable to AMP activation at any given time suggests that additional regulatory interactions, such as compartmentalization or covalent modifications (e.g., phosphorylation), are also involved in this binding process [6].
The tumor suppressor LKB1 is the major upstream serine/threonine kinase that phosphorylates Thr172 in the α subunit and activates the AMPK complex. Of importance is the finding that the LKB1 complex is not directly activated by AMP, but that the latter induces conformational changes in AMPK rendering it a preferable substrate to LKB1 [24]. The human LKB1 gene encodes a single 433-amino acid protein [25], which includes three structural domains: an N-terminal nuclear localization domain, a central catalytic domain and a C-terminal prenylation site that targets the enzyme to membranes [26]. This enzyme plays an important role during embryogenesis and is involved in epithelial, glial and neuronal cell polarity determination [27]. LKB1-knockout mouse embryos die in uterus due to multiple abnormalities, including neural tube and vascular defects [25]. LKB1 is most active in a complex with two accessory proteins: the STE20-related adaptor (STRAD) and the mouse embryo scaffolding protein (MO25) [24, 28, 29]. The pseudokinase STRAD, which binds ATP but lacks a phosphorylation function, enhances the catalytic function of LKB1. MO25, which interacts with the C-terminus of STRAD, stabilizes the entire LKB1 complex [24]. Interestingly, LKB1, like AMPK, shuttles between the nucleus and the cytoplasm [25, 30]. Ribosomal S6 kinase (RSK)- and protein kinase A (PKA)-induced phosphorylation of Ser431 enhances the association of LKB1 with intracellular membranes [31]. It is assumed that this membrane targeting is important to maintain enzymatic activity.
Although AMPK activation is lost when LKB1 expression is silenced, the basal activity of AMPK remains intact [32]. The ability of the AMPK complex to maintain an intrinsic Thr172-autophosphorylating capacity is independent of the fluctuation in AMP levels and seems to maintain a basal intrinsic activity of the complex that allows cells to respond immediately to energetic challenges [33].
Calmodulin-dependent protein kinase kinases (CaMKKs), the upstream kinases for calmodulin-dependent protein kinase I and IV, can also phosphorylate Thr172 and activate AMPK. CaMKKβ, rather than CaMKKα, is the main isoform of the enzyme that activates AMPK [34-36]. Because in many cases intracellular Ca2+ signaling often precedes energy utilization and demand, it is conceivable that CaMKKβ prepares cells for a significant increase of energy demand by activating AMPK [37].
Transforming growth factor-β-activated kinase-1 (TAK1) directly phosphorylates AMPK in yeast [38]. Recently, the mammalian TAK1 homolog (previously known as AMPK-kinase), which also phosphorylates Thr172 in AMPK has been found in various mammalian tissues, including skeletal muscle [38-40]. It has been suggested that tumor necrosis factor-α (TNF-α) and transforming growth factor-β activate TAK1 [41]. In addition, 5-aminoimidazole-4-carboxamide-1β-D-ribofuranoside (AICAR) and the biguanide metformin also activate TAK1. This effect of metformin was lost in mice cardiomyocytes following the disruption of TAK1 expression [42]. Further studies will ascertain the physiological function of TAK1 in cellular energy metabolism. Protein phosphatases 2A and 2C (PP2A and PP2C) inactivate AMPK by dephosphorylating pThr172 [33, 43]. The activity of these enzymes is negatively regulated by AMP and free fatty acids [44].
In summary, changes in energy and calcium metabolism in cells underlie the activation process of AMPK. Among the effectors that act in concert to induce Thr172 phosphorylation, LKB1, CaMKKβ and TAK1 seem to be prominent.
Metabolic Functions of AMPK
AMPK phosphorylates serine moieties in target proteins. It mostly interacts with a serine moiety within a 9-amino acid motif. In human, this motif is Φ-Ψ-X-X-S/T-X-X-X-Φ, where Φ, Ψ and X denote hydrophobic, basic or any other amino acid, respectively [45-47]. Many of these target proteins regulate key metabolic functions, such as glucose uptake, glycolysis, fatty acid oxidation, cholesterol synthesis, glycogen synthesis, gluconeogenesis, protein synthesis or lipolysis [48]. These various functions inhibit anabolic processes and conserve ATP, on one hand, and stimulate catabolic pathways to produce ATP, on the other [46].
Skeletal muscles and heart
Skeletal muscle mass is the main target for insulin-stimulated glucose disposal, whereas insulin resistance is a major contributing factor to the development type 2 diabetes. AMPK-dependent stimulation of glucose transport in skeletal muscles is independent of insulin, phorbol ester or passive stretch [19, 46]. In contrast, muscle contractions, hypoxia, hyperosmolar shock, mitochondrial uncouplers and electron transport inhibitors activate AMPK due to relative energy depletion [49]. Both hypoxia and mitochondrial uncoupling increase the rate of glucose transport in mouse skeletal muscles but fail to produce a similar response in skeletal muscles bearing a dominant-negative AMPKα2 subunit [50, 51].
The activation of muscle AMPK by exogenous compounds or by contraction recruits GLUT-4 to the plasma membrane and augments the rate of glucose transport in a non-insulin-dependent manner [52, 53]. AMPK-induced translocation of GLUT-4-containing vesicles to the plasma membrane is preceded by the phosphorylation of the protein AS-160 at Thr642. This phosphorylated form of AS-160 releases the vesicle from intracellular storages and allows their recruitment to the plasma membrane [54, 55]. In addition, AMPK upregulates the expression of genes encoding GLUT-4 and hexokinase II and stimulates glycogen synthesis in muscles by allosteric activation of glucose-6-phosphatase-induced activity [56-58]. Various studies link the glucose transport stimulatory effect of AMPK in skeletal muscles to the activation of ERK1/2, p38-MAPK, Pyk2, PLD, αPKC and Grb2 [19]. In addition to the GLUT-4 translocation, AMPK also exerts its anabolic function in skeletal muscles by activating two major citric acid cycle enzymes: citrate synthase and succinate dehydrogenase [59-61]. AMPK also stimulates glycolysis in cardiomyocytes (and hepatocyes) by activating 6-phosphofructo-2-kinase (PFK2) [62, 63]. AMPK (predominantly complexes with the α2 isoform) has a cardioprotective role in augmenting glucose transport and glycolysis in ischemic hearts [64, 65]. Increased myocardial ischemia injury due to enhanced post ischemic myocardial apoptosis, extended infarct size and worsened cardiac functional recovery were inflicted in mice bearing a dominant negative AMPKα2 in their cardiomyocytes [66]. In non-insulin-sensitive cells that do not express GLUT-4, AMPK increases glucose uptake possibly by activating the ubiquitous GLUT-1 that resides in the plasma membrane [67].
Although the outcome of insulin action and AMPK activation of the glucose transport system in skeletal muscles is similar, the transduction mechanism employed by insulin to recruit GLUT-4 to the plasma membrane is entirely different and independent of that recruited by AMPK. Indeed, when costimulated, both the AMPK- and the insulin-dependent pathway increase the rate of glucose transport in an additive manner [68]. The interactions between these two main pathways that regulate glucose metabolism in skeletal muscles have been investigated [69]. Disruption of the AMPKα2 function in skeletal muscles in mice resulted in glucose intolerance and insulin resistance, lending more support to the hypothesis that in addition to normal insulin function, the AMPKα2 isoform also plays a role in maintaining normal insulin sensitivity and reactivity of muscles [19]. In cardiac muscle, however, insulin antagonizes the activation of AMPK by an Akt/PKB-dependent phosphorylation of Ser485 or Ser491 in AMPK α1 or α2, respectively, which attenuates the phosphorylation of Thr172 by LKB1 [70-72].
The observation that physical activity induces translocation of AMPKα2 from the cytoplasm to the nucleus supports the idea that AMPK also regulates transcriptional functions, such as the increased expression of GLUT-4 in exercising muscles [73-75]. AMPK phosphorylates histone deacetylase-5 and releases it from a complex with myocyte enhancer factor-2 (MEF2), rendering the latter accessible to threonine residues phosphorylation by p38-MAPK and allowing it to form an active transcription complex of the GLUT-4 gene [76].
The blood glucose lowering effect of exercise in type 2 diabetic patients results from an insulin-independent increased rate of glucose transport and enhanced lipid oxidation in skeletal muscles [58]. Interestingly, exercise and AMPK activation also render skeletal muscles more sensitive to insulin action. Indeed, preincubation of isolated rat skeletal muscles with AICAR enhanced insulin-stimulated glucose transport [77]. However, such effects were not observed in cultured myotubes (C2C12) or primary human muscle cells, in which pretreatment with AICAR did not alter their sensitivity to insulin [78]. AMPK-dependent inactivation of IRS-1 by Ser789 phosphorylation in these cells may explain this phenomenon [79].
It is important to note that contraction-mediated glucose uptake is only partially increased in isolated muscles from AMPKα2 whole body knockout mice [80]. Moreover, the simultaneous silencing of the α1 and α2 subunits in mouse skeletal muscles led to a modest reduction in the rate of glucose uptake in contracting muscles [19]. Such findings support the claim that AMPK is not required for contraction-mediated augmentation of glucose transport in skeletal muscles. In contrast, others have found that inhibition of the AMPK complex with Compound C decreased contraction-stimulated glucose transport in rat skeletal muscles [81]. Therefore, further investigation of the specific role of AMPK in the regulation of the glucose transport system in resting and contracting skeletal muscles during acute exercise or endurance training is required. This may lead to the discovery of new cellular targets for the development of novel drugs that mimic effects of muscle contraction and augment glucose transport in skeletal muscle in non-insulin and non-AMPK-dependent mechanisms.
Reduced AMPK activity in insulin resistance and type 2 diabetes has recently been associated with mitochondrial dysfunction and impaired metabolism of lipids in skeletal muscles [82]. Glucose storage in skeletal muscles serves as an immediate source for ATP production, especially during exercise and short-term fasting. Free fatty acids provide an abundant and long-term stock of energy for peripheral energy demands. Fatty acid oxidation in working muscles produces energy 3-fold greater than comparable carbohydrate resources [83]. Inhibition of acetyl-CoA carboxylase (ACC) by AMPK in skeletal muscles improves β-oxidation of fatty acid in the same mechanism as described below for hepatocytes.
Peroxisome proliferator-activated receptor γ (PPARγ) coactivators-1α and -1β (PGC-1α/β) are critical stimulators of mitochondrial biogenesis in response to various stimuli and stressful conditions, such as different diets or physical activity [84]. Muscle PGC-1α is activated by AMPK and by NAD+-dependent type III deacetylase (SIRT1). It has recently been shown that AMPK indirectly controls SIRT1 activity by increasing NAD+ content. SIRT1, in turn, deacetylases and activates PGC-1α [85, 86]. This introduces a functional crosstalk between two important energy sensors that synchronize energy production in mitochondria.
Adipose tissue
The activation of AMPK in fat tissues leads to decreased lipogenic flux, massive fatty acid oxidation and decreased triglyceride synthesis. Fasting, physical exercise or treatment with β-adrenergic agonists activates AMPK via a cAMP-dependent mechanism. The α1 catalytic subunit is the predominant isoform expressed in adipocytes and is critical for the major effects of the AMPK complex [87]. Not only that AMPK activation in adipocytes just marginally increases glucose uptake, but active AMPK antagonizes the augmenting effects of insulin on GLUT-4-mediated glucose uptake. The mechanism of this phenomenon is not clear, but these findings agree with the view that unlike skeletal muscles, glucose in adipocytes is predominantly utilized anabolically for lipid storage [88].
AMPK also regulates lipolysis in adipocytes by inactivating hormone-sensitive lipase (HSL) by a targeted serine phosphorylation [87, 89]. Normally, receptor-coupled adenylate cyclase increases lipolysis via cAMP-dependent protein kinase-A1, which activates HSL. Interestingly, exposure of adipocytes to AICAR blocks lipolysis, which is induced by this mechanism [90]. Both basal and isoproterenol-stimulated lipolysis were elevated and the antilipolytic effect of AICAR was lost in adipocytes from AMPKα1 knockout mice [91]. It appears that AMPK prevents recycling and release of fatty acids from triglycerides, a process which consumes ATP. AMPKα2 has a direct or indirect role in adipose tissue function since its total deletion in mice resulted in an excessive weight gain upon a high-fat diet, but did not entail glucose intolerance [92].
Activation of AMPK in human adipose tissue leads to an increased expression of adiponectin, which is a potent insulin sensitizer in skeletal muscles [87]. The cAMP response element binding protein (CREB), which is over-activated in adipocytes of obese mice, triggers the expression of the ATF3. The latter is a transcriptional repressor that binds to and inhibits the transcription of the adiponectin and GLUT-4 genes. These interactions have recently been linked to the development of hyperglycemia in type 2 diabetic patients [93]. The peripheral effects of adiponectin may explain the significant contribution of AMPK activating drugs to the prevention and improvement of insulin resistance in obese diabetic patients.
Liver
The main function of AMPK in the liver is to augment fatty acid oxidation and to prevent cholesterol and triglycerides biosynthesis. Liver-specific AMPKα2 deletion in mice enhances hepatic lipogenesis, increases plasma triglyceride levels and hepatic glucose production. Conversely, overexpression of AMPKα2 in hepatocytes decreases plasma triglyceride level [94, 95]. AMPK also reduces mRNA content of the sterol regulatory element binding protein-1 (SREBP-1) [96]. Over-function of this factor has been associated with the increased prevalence of dyslipidemia in type 2 diabetes. Activation of AMPK also reduces the cellular content of the mRNA of the carbohydrate responsive element-binding protein (ChREBP). This factor, otherwise, upregulates lipogenesis and therefore plays a key role in inducing of hyperlipidemia in type 2 diabetes patients [97].
Adipose tissue-derived adiponectin also improves lipid metabolism in the liver of diabetic obese mice by decreasing fatty acid biosynthesis and increasing mitochondrial fatty acid oxidation [98]. The beneficial effects of AMPK in the liver are attenuated when adiponectin synthesis and secretion from adipose tissues is decreased in diabetes and obesity [93].
Type 2 diabetes is characterized by fasting hyperglycemia and impaired peripheral glucose utilization. A key contributing factor to these abnormalities is the failure of insulin to suppress gluconeogenesis and hepatic glucose production. Liver-specific AMPKα2-knockout mice develop hyperglycemia and glucose intolerance and this is associated with an increased hepatic glucose production. Conversely, the stimulation of AMPK in wild type mice dramatically reduces hepatic glucose output [99]. The suppression of gluconeogenesis by AMPK results from the inhibition of the transcription of phosphoenol pyruvate carboxy kinase (PEPCK), the key regulatory gluconeogeneic enzyme [57]. In addition, AMPK attenuates the synthesis of cholesterol and glycogen in hepatocytes by deactivation of HMG-CoA reductase and glycogen synthase, respectively. In addition, AMPK downregulates the expression of enzymes that are centrally involved in fatty acid synthesis and gluconeogenesis by inhibiting the transcription factors SREBP-1c, ChREBP and HNF-4α, and by attenuating the activity of transcriptional coactivators, like p300 and TORC2 [46]. For example, when the latter is phosphorylated it forms a cytoplasmic complex with 14-3-3 protein, which inhibits the transcription of gluconeogenic enzymes [100].
AMPK also phosphorylates and deactivates ACC [101], an enzyme that exists as two isoforms: ACC1 (cytoplasmic) and ACC2 (predominantly mitochondrial) [102]. The inhibition of the former reduces fatty acid synthesis in cells. Malonyl-CoA, the product of ACC2, is a potent blocker of carnitine palmitoyltransferase-1 (CPT1), which transports long chain fatty acids to mitochondria. When ACC2 is inhibited, the flux of these fatty acids to mitochondria and their oxidation is increased. In addition, AMPK directly stimulates free fatty acid uptake to cells by translocating the fatty acid translocase CD38 to the plasma membrane [46].
β-cells
Low glucose levels activate AMPK in β-cells. Overexpression of wild type AMPK or constitutively active AMPK, or its pharmacological activation attenuate glucose-induced insulin secretion, whereas the expression of a dominant-negative AMPK in cultured β-cells increases it [103]. It has been suggested that the biguanide metformin affects insulin secretion in β-cells by activating AMPK [104, 105].
Unlike peripheral tissues like skeletal muscle, which predominantly express the α2 subunit of AMPK, the α1 subunit is more abundant in rodent β-cells than the α2 subunit. The former is found most frequently in the cytoplasm, while the latter is distributed between the cytoplasm and the nuclear compartment. It has been proposed that the AMPKα1 complex participates in the electrochemical activity of the cells, whereas the nuclear AMPKα2 complex is involved in regulating gene transcription. Interestingly, neutralization of the α2 subunit in β-cells enhances the transcription of the preproinsulin gene [106]. Therefore, it seems that the transcriptional regulation of AMPKα2 in β-cells greatly differs from the transcriptional regulation it mediates in skeletal muscles [76].
Recent studies on the role of AMPK in the regulation of insulin secretion in β-cells have associated it with the mTOR pathway, energy availability, protein synthesis, cell growth and apoptosis [69]. Collectively, these results demonstrate the complexity of AMPK function in regulating insulin secretion in β-cells and the need for thorough investigations of the complexity of such direct and/or indirect interactions.
Non-metabolic functions of AMPK
AMPK is involved in numerous cellular and physiological interactions not directly related to metabolic homeostasis or diabetes. It is clear that non-specific and non-tissue/cell targeted AMPK activators for the treatment of diabetes and metabolic disorders may also affect these processes and produce diverse side effects. The following is a short summary of such critical functions of AMPK.
An important cross-talk between AMPK and endothelial NO synthase (eNOS) regulates vascular endothelial cell dilatory functions. The AMPK activity in vascular endothelial cells is controlled directly by AMP and indirectly by nitric oxide, the product of eNOS. Following the activation of soluble guanylate cyclase by NO, the former activates CaMKK, a positive upstream activator of AMPK [107]. In a positive feedback loop, AMPKα1 complex phosphorylates eNOS at Ser1177 in an Akt/PKB-dependent manner and increases NO production [108]. This reciprocal and positive feedback mechanism integrates stressful stimuli (e.g., hypoxia) and metabolic (e.g., hypoglycemia) signals to maintain an adequate circulation in critical organs.
AMPK inhibits DNA replication in various cell lines by causing a G1/S-phase cell cycle arrest. This phenomenon is accompanied with the accumulation of tumor suppressor factor p53 and the cyclin-dependent kinase inhibitors p21 and p27 [46]. Another mechanism that may explain some antiproliferative effects of AMPK is the targeting of the RNA-binding protein HuR to the nucleus, where it interacts with and decreases the half-life of mRNAs encoding p21, cyclin A, and cyclin B1 [109]. AMPK has also been implicated in downregulating protein synthesis in cells by inhibiting mammalian target of rapamycin (mTOR) [79], and by phosphorylating the elongation factor-2 kinase, which than inhibits the elongation step [110]. A rare familial deactivating mutation in LKB1 has been linked to the development of hamartomas in humans (Cowden syndrome, Peutz-Jeghers syndrome and tuberose sclerosis). Despite the abnormal growth, cells in these hamartomas retain a normal differentiated state. Interestingly, some reports claim that diabetic patients treated with metformin have a decreased risk of cancer-related mortality than patients treated with other antidiabetic drugs or insulin. These beneficial effects of metformin have been attributed to the activation of the AMPK pathway [111]. These and other findings have initiated a great interest in the potential antitumorgenic activity of AMPK-targeted drugs [112].
Critical functions of primary immune cells, such as chemotaxis and cytokine secretion, are also regulated by AMPK [113]. In fact, the stimulation of AMPK in macrophages from diabetic mice was reduced in comparison with normal macrophages. This perhaps underlies the impeded functional capacity (i.e., reduced macropinocytosis) of the former cells in diabetes [114].
It has recently been shown that AMPK also participates in the regulation of appetite and food intake. Constitutive activation of AMPK in the mouse hypothalamus increases significantly food uptake and body weight, whereas its inhibition has the opposite effects [115, 116]. Noteworthy are the observations that some drugs and factors have opposite effects on hypothalamic and peripheral AMPK complexes. For example, leptin and metformin activate AMPK in skeletal muscles while inhibiting it in the hypothalamus. In contrast, ghrelin and cannabionoids stimulate AMPK in the hypothalamus, but inhibit it in the liver and adipose tissues [117]. These disparate functions are not well-investigated, but seem to be related to diverse heteromeric structures of AMPK complexes in various regions of the central nervous system and in peripheral tissues.
An interesting relationship has been found between AMPK and the cystic fibrosis transmembranal conductance regulator (CFTR). Both proteins are colocalized in the apical membrane of lung secretory epithelial cells. AMPK phosphorylates CFTR at two serine moieties that maintain the chloride channel closed. Therefore, systemic activation of AMPK may have detrimental effects whereas site-specific pulmonary inhibition of AMPK might be considered therapeutically relevant for the treatment of cystic fibrosis. The potential of such treatments with AMPK disruptors or inhibitors in the lung remains to be elucidated [118].
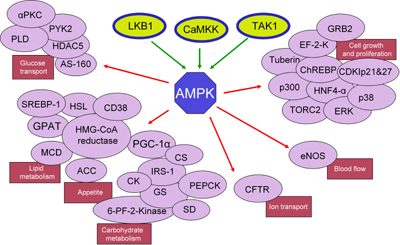
Figure 1 depicts the three main kinases that activate AMPK and its multiple downstream targets, classified according to metabolic, cellular and physiological functions.
In summary, the heterotrimeric structure of the AMPK complex does not only differ among tissues and organs, its activation results in an array of cellular and metabolic responses. Therefore, global activators of AMPK aimed at producing favorable therapeutic effects may exert undesirable side effects due to peripheral and central interactions. This leads to the conclusion that more specific drugs need to be developed. Drugs targeted chemically or by way of administration to specific organs expressing distinct AMPK hetrotrimeric complexes present a potential solution to a rational AMPK-targeted therapy.
 |
 |
Figure 1. Metabolic pathways and functions regulated by AMPK. AMPK can be activated directly by three kinases, LKB1, TAK1 and CaMKK. When activated in various tissues and organs different AMPK complexes mediate a variety of cellular and physiological responses by activating cell-specific targets (e.g., enzymes, transcription factors and docking proteins). Major effects of AMPK activations are metabolic (carbohydrate and lipid metabolism), appetite regulation, cell growth and differentiation, vascular function (blood flow) and basic cellular functions (chloride ion transport). Abbreviations: ACC: acetyl-Co-A carboxylase. AS-160: Akt substrate of 160 kDa. CDKIp21&27: cyclin-dependent kinase inhibitors p21 and p27. CFTR: cystic fibrosis transmembrane conductance regulator. ChREBP: carbohydrate responsive element binding protein. CK: creatine kinase. CS: citrate synthase. EF-2-K: elongation factor 2 kinase. eNOS: endothelial nitric oxide synthase. ERK: extracellular signal-regulated kinases. GPAT: glycerol-3-phosphate acyltransferase. GRB2: growth factor receptor-bound protein 2. GS: glycogen synthase. HDAC25: histone deacetylase 25. HSL: hormone sensitive lipase. HMG-CoA reductase: 3-hydroxy-3-methyl-glutaryl-CoA reductase. HNF4-α: hepatocyte nuclear factor 4α. IRS-1: insulin receptor substrate 1. MCD: malonyl-CoA decarboxylase. p38: p38 mitogen-activated protein kinase. PEPCK: phosphoenolpyruvate carboxy kinase. 6-PF-2-Kinase: 6-phosphofructo-2-kinase. PGC-1α: peroxisome proliferator-activated receptor-γ-coactivator-1α. PYK2: proline-rich tyrosine kinase 2. SD: succinate dehydrogenase. SREBP-1: sterol regulatory element binding protein. TORC2: target of rapamycin complex 2. |
|
Chemical activators of AMPK
The search for novel AMPK activators by rational drug design, screening of vast chemical libraries and testing of various plant extracts has produced numerous reports on new compounds. In the following section, we discuss and classify a collection of such drugs and compounds that activate AMPK. We make the classification according to chemical structures and function.
Metformin and thiazolidinediones
Two groups of common antidiabetic drugs, biguanides and thiazolidinediones (TZD), are believed to mediate part of their effects through AMPK activation. Biguanides, and specifically metformin, inhibit hepatic gluconeogenesis and augment the rate of glucose uptake in skeletal muscles [119]. Some of these effects have been attributed to the activation of AMPKα2 in skeletal muscles. However, the pharmacological significance of this mechanism in diabetic patients is questionable because the required effective concentration of metformin in vitro was as high as 0.5 mM, whereas the plasma effective concentration of the drug in man is at least one order of magnitude lower [96]. Since metformin is excreted in the urine unmetabolized, no active metabolites of this drug can be implicated in its action in skeletal muscles and other tissues [120]. Metformin does not induce LKB1 phosphorylation and activity in skeletal muscles, it fails to increase calcium influx and it does not alter protein phosphatase activity [121]. The main antidiabetic effect of metformin is attributed to hepatic activation of AMPK followed by the inhibition of gluconeogenesis.
Pharmacokinetic data on metformin also supports the hypothesis that the liver is the main target for this drug. Orally administered metformin is effectively absorbed from the gastrointestinal tract to the portal vein. Thus, due to this first-pass effect, the liver is exposed to high concentration of the drug. Therefore, in contrast to skeletal muscle, the effect of metformin in the liver is mediated by LKB1 as the specific hepatic knockout of LKB1 in diabetic mice completely rendered them insensitive to metformin [116]. Of concern is a recent report that links metformin-induced activation of AMPK to an increased biogenesis of Alzheimer's amyloid in mice brains [122]. Therefore, novel AMPK-activating drugs devoid of such serious side effects are needed.
The second class of antidiabetic drugs that stimulate AMPK is thiazolidinediones (TZD). Members of this group that bind to and activate PPARγ improve insulin sensitivity in various peripheral tissues. TZD, such as rosiglitazone and pioglitazone, downregulate lipolysis and reduce the content of fatty acids in adipocytes [123]. These drugs also inhibit the release of several adipokines from adipose cells, such as tumor necrosis factor α (TNF-α), interleukin-6 and resistin, which induce muscle insulin resistance. Concomitantly, TZD augment the secretion of the insulin-sensitizing factor adiponectin from adipose tissue in man and rodents [124]. Adiponectin stimulates glucose uptake and fatty acid oxidation in skeletal muscles and inhibits gluconeogenesis in the liver by activating AMPK [98]. Expression of a dominant negative AMPKα1 mutant in adipocytes or treatments with synthetic inhibitors of AMPK abolished these effects of adiponectin [125]. These findings indicate that AMPK also plays a key role in mediating hepatic metabolic effects of adiponectin.
It has also been reported that the skeletal muscle levels of several oxidative phosphorylation enzymes and of PGC-1 were increased in diabetic patients treated with TZD over 6 months [126]. PGC-1 regulates the expression of several genes that are involved in mitochondrial bioenergetics [127]. Moreover, TZD also increase the expression of super oxide dismutase-2 (SOD2) and quinone oxidoreductase-1 (NQO1), providing an efficient antioxidant defense against elevated levels of reactive oxygen species [128]. Importantly, in vitro experiments suggest that TZD-dependent activation of AMPK-dependent pathways in insulin sensitive tissue is not exclusively mediated by adiponectin. Finally, TZD effects were recorded in adipocytes in which PPARγ expression was blocked [129]. It has recently been reported that the TZD derivative BLX-1002, which does not bind to PPARγ, stimulates insulin secretion from isolated normal and diabetic pancreatic islets or dispersed β-cells only upon incubation under high glucose. Part of this effect has been attributed to the activation of AMPK in β-cells [130].
D-xylose and lipophilic D-xylose derivatives
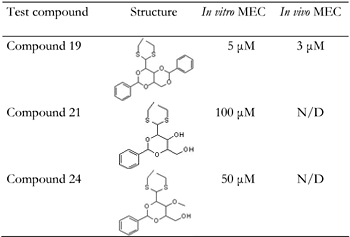
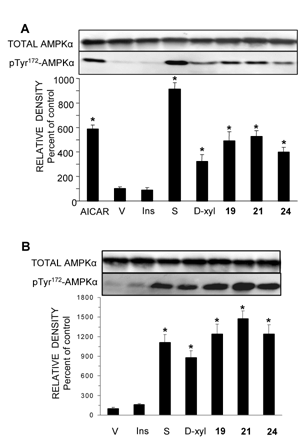
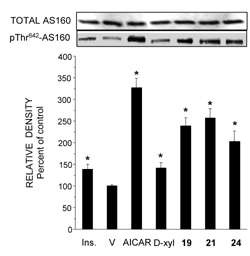
A decade ago Winder et al., introduced AMPK as a target for developing novel drugs for the treatment of type 2 diabetes [131]. Dozens of molecules that activate AMPK, directly or indirectly, were synthesized or extracted from plants. In the course of our work, we found that the pentose D-xylose augmented the rate of glucose transport in L6 and human myotubes under high glucose conditions by activating AMPK [132]. Because this effect of D-xylose was obtained at very high concentrations (10-20 mM), we synthesized more potent lipophilic derivatives of D-xylose. Among these, three highly lipophilic compounds 2,4;3,5-dibenzylidene-D-xylose-diethyl-dithioacetal (Compound 19), 2,4-benzylidene-D-xylose-diethyl-dithioacetal (Compound 21) and 2,4-benzylidene-D-xylose-3-O-methyl-diethyl-dithioacetal (Compound 24) exerted significant glucose transport stimulatory effects at low concentrations (5-100 μM) in rat and human cultured myotubes (Table 1). These effects were decreased in the presence of the AMPK inhibitor Compound C. All three derivatives and the parent compound, D-xylose, induced Thr172 phosphorylation of AMPKα and of Thr642 in the downstream substrate AS-160 (Figure 2 and Figure 3). Our study also shows that the inhibition of the insulin transduction pathway by wortmannin and an AKT inhibitor did not interfere with the glucose transport stimulatory function of these derivatives. The exact mechanism of action of these compounds is still under investigation. Of major importance is the finding that Compound 19 significantly reduced blood glucose levels towards the normoglycemic range in streptozotocin-diabetic mice and in the genetically diabetic KKAy mice. Table 1 depicts the structures of these compounds and their minimal effective concentrations in augmenting the rate of glucose transport in L6 myotubes and in lowering blood glucose levels in streptozotocin-diabetic C57/Black mice. These findings point to the potential of these compounds in the development of novel and long-acting antihyperglycemic compounds.
Table
1.
Structure and function of the D-xylose derivative compounds 19, 21 and 24 |
|
 |
|
Legend:
The table shows the structures of three compounds and their minimal effective concentrations (MEC) required to augment the rate of hexose uptake in cultured L6 myotubes and to reduce the blood glucose levels towards normoglycemia in STZ-diabetic C57/black male mice. Mice were injected subcutaneously twice daily, for 5 days, with 50 mg/kg of each compound dispersed in sesame oil. N/D: not determined. |
|
 |
|
Figure 2. D-Xylose and Compounds 19, 21 and 24 activate AMPK. A: L6 rat myotube cultures were washed and received fresh medium supplemented with 2% (v/v) FCS, 23.0 mM D-glucose supplemented with 20 mM of D-xylose (D-xyl), 5 μM of Compound 19, 150 μM of Compound 21 or 50 μM of Compound 24. These compounds were present in the medium for 40 min, 12 h, 30 min and 2 h, respectively. Control myotubes received the vehicle (V) only. AICAR (4 mM), 100 nM of insulin (Ins) and 0.25 M of D-sorbitol (S) were present for 1h, 20 min and 30 min, respectively. Whole cell lysates were prepared and Western blot analyses were performed with antibodies against AMPKα and pThr172-AMPKα. B: Human myotubes were treated as described above and taken for Western blot analysis of AMPKα and pThr172-AMPKα. Representative blot and a summary of n = 3 (* p < 0.05) in comparison with the respective controls. Reproduced with permission from [135]. |
|
 |
|
Figure 3. D-Xylose and Compounds 19, 21 and 24 activate AS160. Whole cell content of AS160 and pThr642-AS160 was determined by Western blot analysis in samples that were prepared from L6 myotubes, as described in the legend to Figure 1. Representative blot and a summary of n = 3 (*< p < 0.05) in comparison the respective controls. Reproduced with permission from [135]. |
|
These three benzylidene derivatives share some structural similarities to other known activators of AMPK. Figure 4 shows compounds that contain three to five aromatic and non-aromatic rings, connected to each other directly or with short linkers. Some of these rings contain heteroatoms, such as oxygen and nitrogen. For example, berberine, an isoquinoline alkaloid derived from Hydrastic Canadensis, and its synthetic derivative dihydroberberine are claimed to be effective antilipogenic and hypoglycemic agents in rodents [133, 134]. Some studies have shown that both are indirect activators of AMPK, probably by inhibiting complex 1 in mitochondria [134, 135]. The furancarboxylic acid derivative compound D942 has also been found to activate AMPK indirectly, due to its ability to bind to NAD(P)H dehydrogenase and attenuate the function of complex 1 [136].
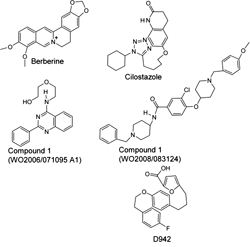 |
|
Figure 4. Molecular structures of AMPK activators sharing structural similarities to the D-xylose derivatives, Compounds 19, 21 and 24. |
|
Cilostazol
Cilostazol is a selective inhibitor of phosphodiesterase-3 (PDE3) that shares structural similarities with the compounds shown in Table 1 and Figure 4. One of the striking effects of this compound is the enhancement of Thr172 phosphorylation and activity of AMPK in human umbilical vein endothelial cells. This activation is followed by downstream phosphorylations of ACC and eNOS. Cilostazol restores vascular endothelial cell function in diabetic rats by PDE3 inhibition [137]. In a study on the effects of cilostazol on thrombospondin-1 expression in hearts of streptozotocin-diabetic rats, the investigators reported that a 4-week treatment reduced blood glucose levels significantly from 22.1 to 16.7 mM [138]. Others have shown that cilostazol ameliorates metabolic abnormalities in diabetic mice or rats via activation of PPARγ and the suppression of inflammatory markers [139]. This drug also improved arterial compliance in patients with peripheral arterial disease while also improving their lipid profile [140]. Further clinical observations and trials will determine the efficacy of such auxiliary antidiabetic effects this drug may possess.
Recently, Zhou et al. reported the structure of two patented polycyclic activators of AMPK that may belong to the group presented in Figure 4 [141]. Compound 1 (patent WO2006/071095) increased AMPK activity 6-fold via an unknown mechanism [142]. Compound 1 (patent WO2008/083124) activated AMPK indirectly. It has been suggested that this effect is mediated via adiponectin receptors. It remains to be investigated whether these compounds also affect the mitochondrial complex 1 [143].
Phytoestrogens
It has been assumed that phytoestrogens could reduce diabetic complications by improving glucose homeostasis and insulin resistance. For example, antilipogenic effects of the phytoestrogen genistein (Figure 5) and its capacity to decrease adiposity were related to the activation of AMPK [144]. Genistein, quercetin, isoginkgetin and epigallocathechin-3-gallate (Figure 5), contain isoflavone and isoflavone-like moieties in their structures. They activate AMPK in 3T3-L1 preadipocytes and adipocytes, adiposarcoma cells and primary mouse hepatocytes, respectively [145]. The activation of AMPK by epigallocathechin-3-gallate seems to be mediated by CaMKK [146]. Interestingly, the antioxidant properties of the gallic acid polyphenol moiety in epigallocathechin-3-gallate may also contribute to the antidiabetic effects of this compound [147]. The mechanism of AMPK activation of the other compounds is not yet clear.
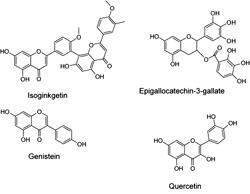 |
|
Figure 5. Molecular structures of phytoestrogens that interact with AMPK. |
|
Momordicosides
Momordicosides, such as maslinic acid, cucurbitane and ginsenosides represent another class of natural compounds that activate AMPK and may also possess antidiabetic properties (Figure 6). The main natural source of maslinic acid is olive's skin; the rest are extracted from Momordica charantia (bitter melon) [148, 149]. In addition to the four ring triterpenoidic structure, the majority of momordicosides contain one or more sugar moieties bound to the ring structure via glycoside bonds. The most abundant sugar moieties are β-D-glucopyranoside, β-D-allopyranoside and β-D-xylopyranoside. Several studies indicate that these compounds increased the rate of glucose transport and induce GLUT-4 translocation to the plasma membrane in L6 or C2C12 myotubes and in 3T3-L1 adipocytes by activating AMPK. When tested in vivo, some momordicosides enhanced fatty acid oxidation and glucose disposal during glucose tolerance tests in insulin-sensitive or insulin-resistant mice or augmented glucose-stimulated insulin secretion in mice [149, 150]. In vitro studies suggest that ginsenosides increase AMPK phosphorylation and activity in 3T3-L1 cells; however, the relevant molecular interactions have not yet been ascertained [151].
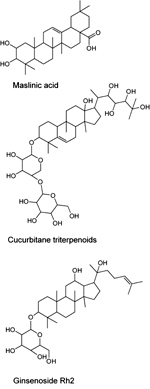 |
|
Figure 6. Molecular structures of momordicosides representing a class of natural compounds that activate AMPK. |
|
Capsaicinoids
Another large group of AMPK activators have one or two phenol, polyphenol or phenolmethyl ether moieties in their structures (Figure 7). The majority of these compounds have been extracted from plants. For example, capsaicinoids, a group of the natural compounds extracted from hot peppers, have been reported to promote fatty acid oxidation and decrease body fat accumulation in diabetic mice. The synthetic structural ester isomer isodihydrocapsiate activates LKB1 both in vitro and in vivo [152]. This substance also increases glucose uptake in L6 myotubes and when administered orally to diabetic mice it substantially reduces blood glucose. A recent report indicated that salidroside, the active ingredient purified from Rhodiola Rosea, stimulated glucose transport and enhanced insulin sensitivity in L6 myotubes and 3T3-L1 adipocytes. The inhibition of AMPK activity by compound C completely abolished these effects of salidroside in L6 myotubes. This suggests that this compound exerts its effects by activating AMPK [153]. Another member of this group is the antioxidant resveratrol, which increases insulin sensitivity and lowers lipids in blood of diabetic and obese mice, most likely by activating SIRT1 [154]. Several natural analogues of resveratrol also activate AMPK [155]. One derivative, combretastatin A-4, a natural cis-stilbene is isolated from the plant Combretum caffrum. It is the most effective and potent member of this group of compounds in downregulating the expression of gluconeogenic enzymes in the liver and reducing the fasting blood glucose level in diabetic mice. The effect of combretastatin A-4 on AMPK is indirect and seems to result from the inhibition of the mitochondrial respiratory chain and a subsequent reduction in ATP levels [156].
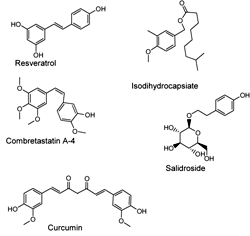 |
|
Figure 7. Molecular structures of compounds that have one or two phenol, polyphenol or phenolmethyl ether moieties. |
|
Curcumin (diferuloylmethane), a polyphenol natural product of the plant Curcuma longa, has attracted significant attention and is undergoing early clinical trials as a novel anticancer agent [157]. It has been shown that curcumin-induced death of ovarian cancer cells is mediated by activating AMPK [158]. In other studies, curcumin rescued isolated mouse pancreatic islets from cytokine-induced death in vitro and prevented streptozotocin-induced diabetes in vivo [159]. Of particular interest is the finding that curcumin-induced activation of AMPK underlies the reduction in hepatic glucose production [160].
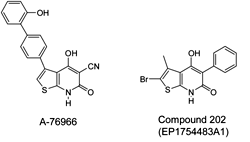
The thienopyridone derivative A769662 possesses antidiabetic effects by direct activation of AMPK [161] (Figure 8). It has been proposed that this compound activates AMPK in an AMP-independent manner, by binding to an alternative allosteric site in the AMPK complex [162]. Others have shown that A769662 binds to the carbohydrate-binding moiety in the γ subunit of the complex and to several amino acid moieties of the β1 subunit, none of which participates in AMP-binding [14]. Noteworthy is the finding that A769662 activates AMPK complexes that exclusively contain the β1 subunit. This property limits the potential therapeutic use of this compound to tissues expressing the ubiquitous β1 subunit, but not to tissue like skeletal muscle that assemble AMPKβ2 complexes. Therefore, the main target of A769662 is most likely the liver in which an activation of β1-containing AMPK complex reduces the expression of gluconeogenic enzymes and hepatic liver production [161]. Another patented thienopyridone derivative recently reported by Zhou et al. is Compound 202 (patent EP1754483A1) [141, 163] (Figure 8). Like A769662, this molecule, which contains a thienopyridone moiety, activates AMPK most likely by interacting with the β1 subunits.
 |
|
Figure 8. Molecular structure of the thienopyridone derivative A769662 and Compound 202. |
|
Furanothiazolidine
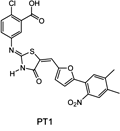
While screening chemical libraries for direct activators of the α-subunit of AMPK Pang et al. [8] discovered a furanothiazolidine derivative, PT1 (Figure 9). When tested in hepatoma HepG2 cells it lowered their lipid content and activated AMPK in a dose-dependent manner. It interacted with the autoinhibitory domain in the α1 subunit and converted AMPK to a constitutively active complex [8]. Experiments with L6 myotubes showed that PT1 effectively increased the phosphorylation of AMPK with no apparent changes in the ATP/ADP ratio in the cells. Moreover, this effect was not mediated via LKB1 because it also occurred in the LKB-deficient cells. PT1-induced phosphorylation of AMPK was lost in cells co-treated with STO-609, a specific CaMKKβ inhibitor, suggesting a critical role for Ca2+-dependent CaMKKβ activation in this process. In vivo tests with this compound are required to ascertain its potential as a prototype molecule for the development of novel antidiabetic drugs.
 |
|
Figure 9. Molecular structure of the furanothiazolidine derivative, PT1, that interacts with the α-subunit of the AMPK complex. |
|
AICAR
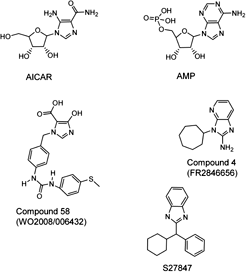
AMP is the natural endogenous activator of the AMPK complex. As explained above, two pairs of AMP molecules interact with two Bateman domains in the γ subunit and allosterically activate the entire AMPK complex. Figure 10 shows AMP and structurally related compounds that induce similar effects. AICAR is metabolized intracellularly to ZMP (5-aminoimidazole-4-carboxamide-1-β-D-ribofuranotide) by adenosine kinase. ZMP is an AMP analog that interacts with Bateman domains in the γ subunit of AMPK and induces allosteric changes in AMPK conformation, allowing kinase activation [52]. Some researchers, however, have claimed that ZMP can also regulate glucose metabolism by direct inhibition of fructose-1,6-biphosphatase in hepatocytes, thereby blocking gluconeogenesis independently of the activation of AMPK [164]. AICAR was tested in animal models of diabetes and exhibited antidiabetic effects as many pathological metabolic parameters in these animals such as, blood glucose level, lipids profile, hepatic glucose output and glucose disposal were improved [165]. AICAR infusion in both healthy and diabetic individuals increased skeletal muscle glucose uptake [166]. It also dramatically reduced the formation of reactive oxygen species in blood and preserved normal endothelial cell function in diabetic patients [167]. In another study, AICAR increased glycogen content in rat skeletal muscles, mostly due to an increased influx of glucose [73].
 |
|
Figure 10. Molecular structures of AMP and structurally related compounds that induce allosterical activation of the AMPK complex. |
|
AICAR also negatively regulates IL-6 and IL-8 gene expression and their secretion from rat adipocytes and skeletal muscle cells. These two proinflammatory mediators are secreted from adipose tissues and play an important role in the etiology of insulin resistance in obese patients. Thus, these effects of AICAR led to the idea that it may serve as a potent drug for increasing insulin sensitivity in peripheral tissues [168]. Nevertheless, AICAR suffers from unfavorable pharmacokinetic properties (i.e., high effective concentration, poor bioavailability and short half life) and severe metabolic complications (e.g., lactic acidosis and massive uric acid production) [169].
Another serious shortcoming of AICAR is the lack of a strict specificity for AMPK. In fact, AICAR interacts with other enzymes, such as S-adenosylhomocysteine hydrolase and glycogen phosphorylase [170]. To solve these problems many groups have tried to develop more potent derivatives of AICAR. One example is the imidazo[4, 5-b]pyridine derivative S27847, which increased the activity of AMPK 7-fold in hepatocytes at micromolar concentrations (Figure 10) [171]. Other structurally related imidazol analogues of AICAR and S27847 are the patented Compound 4 and Compound 58 [141, 172, 173]. These derivatives are less effective than S27847; at 500 and 200 μM they increased AMPK activity 3- and 2-fold, respectively, in comparison with the control treatments.
Other structurally unrelated compounds
Chromium picolinate
Some other molecules that activate AMPK do not belong to the various classes of chemically and structurally related compounds described above. One such compound is chromium picolinate, which is used as an antidiabetic food supplement. Recent reports suggest that some antidiabetic effects of this compound result from AMPK activation and the inhibition of resistin secretion from adipocytes. Resistin is a 12.5 kDa cysteine-rich adipokine known to induce insulin resistance in rodents [174]. Such effects are not found in man due to the lack of resistin expression and secretion [175]. Therefore, this resistin-dependent mechanism is not involved in the antidiabetic effects of chromium picolinate in man.
α-lipoic acid
α-lipoic acid decreases lipid accumulation in rat skeletal muscle and steatosis in the obese rat liver by augmenting AMPK phosphorylation [176]. It has been recently shown that an incubation of C2C12 myotubes with α-lipoic acid increased the activity and Thr172 phosphorylation of the AMPKα2 subunit, followed by Ser79 phosphorylation in ACC. Furthermore, inhibition of CaMKK with the selective inhibitor STO-609 abolished α-lipoic acid-stimulated AMPK activation, with a concomitant reduction of Ser79 phosphorylation in ACC. When short interfering-RNA against CaMKK was used to silence its expression, it abolished α-lipoic acid-induced AMPK activation. These data indicate that CaMKK is possibly the target for α-lipoic acid-induced AMPK activation in myotubes [177].
Kainic acid
Another interesting molecule that activates AMPK in the brain is kainic acid (extracted from various seaweeds) [178]. Of interest are the reports showing specific receptors for kainic acid in the brain. It remains to be investigated whether kainic acid affects energy metabolism in the brain and possibly some important metabolic functions, like appetite.
Cannabinoids
Cannabinoids interact with AMPK complexes in various tissues in opposing manners. They activate AMPK in the hypothalamus and the heart, and inhibit it in the liver and adipose tissues. It has been assumed that cardioprotective effects of cannabinoids result from activating AMPK-dependent metabolic pathways [179]. Nevertheless, no significant benefits of these compounds in terms of blood glucose normalization in diabetes have yet been reported.
Long chain-fatty acids
It has also been proposed that long chain-fatty acids activate AMPK [180]. However, these effects are short-lived and transient due to efficient β-oxidation. Notwithstanding, such effects of long chain fatty acid have been documented in rats and mice and isolated insulin-sensitive tissues. Substituted α,ω-dicarboxylic acids of 14-18C fatty acids (MEDICA analogs), which are not metabolized beyond their acyl-CoA thioesters, also activate recombinant AMPK in a free cell system, in cell lines such as HepG2 and 3T3-L1 and in diabetic db/db mice. Activation of AMPK in the latter animal model normalized blood glucose and suppressed hepatic glucose production [180]. On interest is the recent finding that oral administration of the short chain fatty acid butyric acid also activated AMPK in skeletal muscles and brown adipocytes of dietary-obese C57 black mice. This treatment also prevented the development of insulin resistance and obesity, decreased blood glucose level and reduced body fat content in the treated animals [181]. Thus, both long and short chain fatty acid may activate AMPK.
Reactive oxygen species
Reactive oxygen species (ROS) such as peroxynitrite (ONOO-) have been associated with the activation of AMPK in cells, especially in tissues with a high oxygen demand [182]. Pathologically relevant concentrations of peroxynitrite are capable of activating AMPK independently of variations in the AMP/ATP ratio. It has been suggested that this process is significantly enhanced following hypoxia and reoxygenation in the heart [183]. Similar AMPK stimulatory activity has also been assigned to the free radical hydrogen peroxide [182]. The idea that activated AMPK prevents oxidative stress associated with diabetes by upregulating mitochondrial uncoupling protein-2 (UCP-2) presents an attractive pathway for antidiabetic effects of such AMPK activators [167]. Similarly, the finding that nitric oxide (NO) per se activates AMPK via the Ca2+-dependent CaMKK pathway in vascular endothelial cells points to another mechanism for AMPK activation, especially in cells and tissues where this radical generation is increased due to an of induction of NOS isotypes.
Leptin
Leptin is a 16 kDa peptide that is secreted from adipocytes and regulates energy metabolism and satiety. Several reports show that it activates AMPK in skeletal muscles but inhibits its activation in the hypothalamus. These disparate tissue-specific effects are remarkably related to the overall metabolic regulation in the organism: activation of AMPK in skeletal muscles potentiates the rates of glucose transport and fatty acid oxidation, whereas in the hypothalamus, the inhibition of AMPK reduces the appetite and food intake. Leptin-overexpressing mice exhibit reduced tissue triacylglycerol content and an increased energy expenditure, due to the peripheral phosphorylation of AMPKα2 isoform [184, 185]. This finding was confirmed when leptin infusion increased AMPKα2 activity in skeletal muscles of control and STZ-diabetic mice [184]. The molecular mechanism of leptin-induced activation of AMPK is not yet clear.
Ghrelin
Ghrelin has the opposite effects of leptin on AMPK. It is secreted from the gastric mucosa and acts as a growth hormone-releasing- and an appetite stimulating factor. Several studies have found that ghrelin activates AMPK in the hypothalamus and in the heart and inhibits it in the liver and adipose tissues [186]. It seems to lack any effect on AMPK phosphorylation in skeletal muscles [187]. The upstream kinase by which ghrelin exerts its effects on AMPK has not yet been identified. However, ghrelin has been shown to induce CaMKKβ-dependent signaling in hypothalamic neurons [188].
Interleukin-6
Interleukin-6 (IL-6) rapidly and markedly increases AMPK activity in skeletal muscles. It also increases fatty acid oxidation and basal and insulin-stimulated glucose uptake by translocation of glucose transporter-4 to the plasma membrane of L6 myotubes. These effects are lost in L6 myotubes overexpressing the dominant negative form of AMPKα2 [189]. The relevance of these finding to diabetes treatment is still under investigation.
AMPK inhibitors
Iodotubercidin and arabinose-adenosine (AraA)
The inhibitor iodotubercidin is a synthetic adenosine derivative that has been used to inhibit AMPK in vitro. It should be noted that this compound also interacts with and inhibits other enzymes, such as glycogen synthase, phosphokinase A, phosphokinase C or casein kinase [190-193].
Arabinose-adenosine (AraA) was originally isolated from the Caribbean sponge Tehya crypta. This compound, which has some antiviral activity, also inhibits various kinases including AMPK [194]. Another shortcoming of these two inhibitors is their inability to inhibit contraction-induced AMPK activation in skeletal muscles [195]. Thus, results on AMPK inhibition that were obtained from experiments with these inhibitors should be reevaluated.
Compound C
The synthetic Compound C is a reversible and an AMP-competitive inhibitor of AMPK [196]. When tested in cells, it fails to completely block all AMPK-dependent stimuli. For instance, while AICAR-dependent activation of AMPK in skeletal muscles was blocked by Compound C, the dinitrophenol-induced activation of AMPK was not affected by it. Moreover, some reports claim that Compound C inhibits adenosine transport into the cells. Thus, when presented together with AICAR it can limit the influx of the latter and therefore prevent its potential to activate AMPK intracellularly [196]. An additional limitation of Compound C is its inconsistent level of inhibition of AMPK activity in different tissues: for example, it is very effective in hepatocytes but much less so in skeletal muscles [81]. This may indicate an AMPK-subunit specificity of this inhibitor. Recent studies show that Compound C also inhibits other protein kinases, such as, ERK1/2 and PHK. Therefore, cellular effects mediated by Compound C should be carefully studied and evaluated.
Conclusions
New compounds that activate AMPK in a complex- and tissue-specific manner may eventually become novel antidiabetic and antiobesity drugs. The ideal AMPK activator should have several properties. It should specifically activate AMPK at a much lower concentration than AICAR. The activation should be targeted to AMPK subunits specifically expressed in the liver, adipocytes or skeletal muscles, but not in the central nervous centers where AMPK activation increases appetite and food consumption. Obviously, oral formulation of drugs that produce minimal side effects is essential.
This review describes some classes of direct and indirect activators of AMPK. Further studies are required to understand the molecular interactions of these compounds and structural requirements for their specificity to the various subunits of AMPK or its upstream effectors. Such ongoing efforts may provide a novel class of drugs to treat diabetes and related metabolic abnormalities in the future.
Conflict of interest statement: The authors declare that they have no competing conflict of interests with respect to financial or other issues.
Acknowledgments:
This work was partially supported by a grant from the Alex Grass Center for Drug Design and Synthesis of Novel Therapeutics (School of Pharmacy, Faculty of Medicine, the Hebrew University of Jerusalem), The Yedidut Foundation (Mexico), and DIAB R & D, France. S. Sasson is a member of the David R. Bloom Center for Pharmacy at the Hebrew University of Jerusalem.
References
- Hardie DG, Scott JW, Pan DA, Hudson ER. Management of cellular energy by the AMP-activated protein kinase system. FEBS Lett 2003. 546(1):113-120. [DOD] [CrossRef]
- Wojtaszewski JF, Birk JB, Frosig C, Holten M, Pilegaard H, Dela F. 5'AMP activated protein kinase expression in human skeletal muscle: effects of strength training and type 2 diabetes. J Physiol 2005. 564(Pt 2):563-573. [DOD] [CrossRef]
- Birk JB, Wojtaszewski JF. Predominant alpha2/beta2/gamma3 AMPK activation during exercise in human skeletal muscle. J Physiol 2006. 577(Pt 3):1021-1032. [DOD] [CrossRef]
- Thornton C, Snowden MA, Carling D. Identification of a novel AMP-activated protein kinase beta subunit isoform that is highly expressed in skeletal muscle. J Biol Chem 1998. 273(20):12443-12450. [DOD] [CrossRef]
- Amodeo GA, Rudolph MJ, Tong L. Crystal structure of the heterotrimer core of Saccharomyces cerevisiae AMPK homologue SNF1. Nature 2007. 449(7161):492-495. [DOD] [CrossRef]
- Xiao B, Heath R, Saiu P, Leiper FC, Leone P, Jing C, Walker PA, Haire L, Eccleston JF, Davis CT, Martin SR, Carling D, Gamblin SJ. Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature 2007. 449(7161):496-500. [DOD] [CrossRef]
- Woods A, Munday MR, Scott J, Yang X, Carlson M, Carling D. Yeast SNF1 is functionally related to mammalian AMP-activated protein kinase and regulates acetyl-CoA carboxylase in vivo. J Biol Chem 1994. 269(30):19509-19515. [DOD]
- Pang T, Zhang ZS, Gu M, Qiu BY, Yu LF, Cao PR, Shao W, Su MB, Li JY, Nan FJ, Li J. Small molecule antagonizes autoinhibition and activates AMP-activated protein kinase in cells. J Biol Chem 2008. 283(23):16051-16060. [DOD] [CrossRef]
- Woods A, Salt I, Scott J, Hardie DG, Carling D. The alpha1 and alpha2 isoforms of the AMP-activated protein kinase have similar activities in rat liver but exhibit differences in substrate specificity in vitro. FEBS Lett 1996. 397(2-3):347-351. [DOD] [CrossRef]
- Salt I, Celler JW, Hawley SA, Prescott A, Woods A, Carling D, Hardie DG. AMP-activated protein kinase: greater AMP dependence, and preferential nuclear localization, of complexes containing the alpha2 isoform. Biochem J 1998. 334(Pt 1):177-187. [DOD]
- Kodiha M, Rassi JG, Brown CM, Stochaj U. Localization of AMP kinase is regulated by stress, cell density, and signaling through the MEK-->ERK1/2 pathway. Am J Physiol Cell Physiol 2007. 293(5):C1427-C1436. [DOD] [CrossRef]
- Hudson ER, Pan DA, James J, Lucocq JM, Hawley SA, Green KA, Baba O, Terashima T, Hardie DG. A novel domain in AMP-activated protein kinase causes glycogen storage bodies similar to those seen in hereditary cardiac arrhythmias. Curr Biol 2003.13(10):861-866. [DOD] [CrossRef]
- Warden SM, Richardson C, O'Donnell J Jr, Stapleton D, Kemp BE, Witters LA. Post-translational modifications of the beta-1 subunit of AMP-activated protein kinase affect enzyme activity and cellular localization. Biochem J 2001. 354(Pt 2):275-283. [DOD] [CrossRef]
- Scott JW, van Denderen BJ, Jorgensen SB, Honeyman JE, Steinberg GR, Oakhill JS, Iseli TJ, Koay A, Gooley PR, Stapleton D, Kemp BE. Thienopyridone drugs are selective activators of AMP-activated protein kinase beta1-containing complexes. Chem Biol 2008. 15(11):1220-1230. [DOD] [CrossRef]
- Bateman A. The structure of a domain common to archaebacteria and the homocystinuria disease protein. Trends Biochem Sci 1997. 22(1):12-13. [DOD] [CrossRef]
- Scott JW, Hawley SA, Green KA, Anis M, Stewart G, Scullion GA, Norman DG, Hardie DG. CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J Clin Invest 2004. 113(2):274-284. [DOD]
- Arad M, Seidman CE, Seidman JG. AMP-activated protein kinase in the heart: role during health and disease. Circ Res 2007. 100(4):474-488. [DOD] [CrossRef]
- Cheung PC, Salt IP, Davies SP, Hardie DG, Carling D. Characterization of AMP-activated protein kinase gamma-subunit isoforms and their role in AMP binding. Biochem J 2000. 346(Pt 3):659-669. [DOD] [CrossRef]
- Fujii N, Jessen N, Goodyear LJ. AMP-activated protein kinase and the regulation of glucose transport. Am J Physiol Endocrinol Metab 2006. 291(5):E867-E877. [DOD] [CrossRef]
- Misra P, Chakrabarti R. The role of AMP kinase in diabetes. Indian J Med Res 2007. 125(3):389-398. [DOD]
- Hardie DG, Salt IP, Hawley SA, Davies SP. AMP-activated protein kinase: an ultrasensitive system for monitoring cellular energy charge. Biochem J 1999. 338 (Pt 3):717-722. [DOD] [CrossRef]
- Hawley SA, Davison M, Woods A, Davies SP, Beri RK, Carling D, Hardie DG. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J Biol Chem 1996. 271(44):27879-27887. [DOD] [CrossRef]
- Hardie DG, Hawley SA. AMP-activated protein kinase: the energy charge hypothesis revisited. Bioessays 2001. 23(12):1112-1119. [DOD] [CrossRef]
- Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, Alessi DR, Hardie DG. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol 2003. 2(4):28. [DOD] [CrossRef]
- Alessi DR, Sakamoto K, Bayascas JR. LKB1-dependent signaling pathways. Annu Rev Biochem 2006. 75:137-163. [DOD] [CrossRef]
- Sapkota GP, Boudeau J, Deak M, Kieloch A, Morrice N, Alessi DR. Identification and characterization of four novel phosphorylation sites (Ser31, Ser325, Thr336 and Thr366) on LKB1/STK11, the protein kinase mutated in Peutz-Jeghers cancer syndrome. Biochem J 2002. 362(Pt 2):481-490. [DOD] [CrossRef]
- Forcet C, Billaud M. Dialogue between LKB1 and AMPK: a hot topic at the cellular pole. Sci STKE 2007. 2007(404):pe51. [DOD] [CrossRef]
- Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A 2004. 101(10):3329-3335. [DOD] [CrossRef]
- Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, Schlattner U, Wallimann T, Carlson M, Carling D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol 2003. 13(22):2004-2008. [DOD] [CrossRef]
- Leff T. AMP-activated protein kinase regulates gene expression by direct phosphorylation of nuclear proteins. Biochem Soc Trans 2003. 31(Pt 1):224-227. [DOD]
- Yoo LI, Chung DC, Yuan J. LKB1--a master tumour suppressor of the small intestine and beyond. Nat Rev Cancer 2002. 2(7):529-535. [DOD] [CrossRef]
- Hawley SA, Selbert MA, Goldstein EG, Edelman AM, Carling D, Hardie DG. 5'-AMP activates the AMP-activated protein kinase cascade, and Ca2+/calmodulin activates the calmodulin-dependent protein kinase I cascade, via three independent mechanisms. J Biol Chem 1995. 270(45):27186-27191. [DOD] [CrossRef]
- Suter M, Riek U, Tuerk R, Schlattner U, Wallimann T, Neumann D. Dissecting the role of 5'-AMP for allosteric stimulation, activation, and deactivation of AMP-activated protein kinase. J Biol Chem 2006. 281(43):32207-32216. [DOD] [CrossRef]
- Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab 2005. 2(1):9-19. [DOD] [CrossRef]
- Hurley RL, Anderson KA, Franzone JM, Kemp BE, Means AR, Witters LA. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J Biol Chem 2005. 280(32):29060-29066. [DOD] [CrossRef]
- Woods A, Dickerson K, Heath R, Hong SP, Momcilovic M, Johnstone SR, Carlson M, Carling D. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab 2005. 2(1):21-33. [DOD] [CrossRef]
- Carling D, Sanders MJ, Woods A. The regulation of AMP-activated protein kinase by upstream kinases. Int J Obes (Lond) 2008. 32(Suppl 4):S55-S59. [DOD] [CrossRef]
- Momcilovic M, Hong SP, Carlson M. Mammalian TAK1 activates Snf1 protein kinase in yeast and phosphorylates AMP-activated protein kinase in vitro. J Biol Chem 2006. 281(35):25336-25343. [DOD] [CrossRef]
- McGee SL, Mustard KJ, Hardie DG, Baar K. Normal hypertrophy accompanied by phosphoryation and activation of AMP-activated protein kinase alpha1 following overload in LKB1 knockout mice. J Physiol 2008. 586(6):1731-1741. [DOD] [CrossRef]
- Shibuya H, Yamaguchi K, Shirakabe K, Tonegawa A, Gotoh Y, Ueno N, Irie K, Nishida E, Matsumoto K. TAB1: an activator of the TAK1 MAPKKK in TGF-beta signal transduction. Science 1996. 272(5265):1179-1182. [DOD] [CrossRef]
- Srivastava AK, Qin X, Wedhas N, Arnush M, Linkhart TA, Chadwick RB, Kumar A. Tumor necrosis factor-alpha augments matrix metalloproteinase-9 production in skeletal muscle cells through the activation of transforming growth factor-beta-activated kinase 1 (TAK1)-dependent signaling pathway. J Biol Chem 2007. 282(48):35113-35124. [DOD] [CrossRef]
- Xie M, Zhang D, Dyck JR, Li Y, Zhang H, Morishima M, Mann DL, Taffet GE, Baldini A, Khoury DS, Schneider MD. A pivotal role for endogenous TGF-beta-activated kinase-1 in the LKB1/AMP-activated protein kinase energy-sensor pathway. Proc Natl Acad Sci U S A 2006. 103(46):17378-17383. [DOD] [CrossRef]
- Davies SP, Helps NR, Cohen PT, Hardie DG. 5'-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2C alpha and native bovine protein phosphatase-2AC. FEBS Lett 1995. 377(3):421-425. [DOD] [CrossRef]
- Wu Y, Song P, Xu J, Zhang M, Zou MH. Activation of protein phosphatase 2A by palmitate inhibits AMP-activated protein kinase. J Biol Chem 2007. 282(13):9777-9788. [DOD] [CrossRef]
- Dale S, Wilson WA, Edelman AM, Hardie DG. Similar substrate recognition motifs for mammalian AMP-activated protein kinase, higher plant HMG-CoA reductase kinase-A, yeast SNF1, and mammalian calmodulin-dependent protein kinase I. FEBS Lett 1995. 361(2-3):191-195. [DOD] [CrossRef]
- Hardie DG. AMP-activated protein kinase as a drug target. Annu Rev Pharmacol Toxicol 2007. 47:185-210. [DOD] [CrossRef]
- Weekes J, Ball KL, Caudwell FB, Hardie DG. Specificity determinants for the AMP-activated protein kinase and its plant homologue analysed using synthetic peptides. FEBS Lett 1993. 334(3):335-339. [DOD] [CrossRef]
- Witczak CA, Sharoff CG, Goodyear LJ. AMP-activated protein kinase in skeletal muscle: from structure and localization to its role as a master regulator of cellular metabolism. Cell Mol Life Sci 2008. 65(23):3737-3755. [DOD] [CrossRef]
- Hayashi T, Hirshman MF, Fujii N, Habinowski SA, Witters LA, Goodyear LJ. Metabolic stress and altered glucose transport: activation of AMP-activated protein kinase as a unifying coupling mechanism. Diabetes 2000. 49(4):527-531. [DOD] [CrossRef]
- Fujii N, Hirshman MF, Kane EM, Ho RC, Peter LE, Seifert MM, Goodyear LJ. AMP-activated protein kinase alpha2 activity is not essential for contraction- and hyperosmolarity-induced glucose transport in skeletal muscle. J Biol Chem 2005. 280(47):39033-39041. [DOD] [CrossRef]
- Halseth AE, Ensor NJ, White TA, Ross SA, Gulve EA. Acute and chronic treatment of ob/ob and db/db mice with AICAR decreases blood glucose concentrations. Biochem Biophys Res Commun 2002. 294(4):798-805. [DOD] [CrossRef]
- Merrill GF, Kurth EJ, Hardie DG, Winder WW. AICA riboside increases AMP-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. Am J Physiol 1997. 273(6 Pt 1):E1107-E1112. [DOD]
- Mu J, Barton ER, Birnbaum MJ. Selective suppression of AMP-activated protein kinase in skeletal muscle: update on 'lazy mice'. Biochem Soc Trans 2003. 31(Pt 1):236-241. [DOD]
- Eguez L, Lee A, Chavez JA, Miinea CP, Kane S, Lienhard GE, McGraw TE. Full intracellular retention of GLUT4 requires AS160 Rab GTPase activating protein. Cell Metab 2005. 2(4):263-272. [DOD] [CrossRef]
- Miinea CP, Sano H, Kane S, Sano E, Fukuda M, Peranen J, Lane WS, Lienhard GE. AS160, the Akt substrate regulating GLUT4 translocation, has a functional Rab GTPase-activating protein domain. Biochem J 2005. 391(Pt 1):87-93. [DOD]
- Barnes BR, Long YC, Steiler TL, Leng Y, Galuska D, Wojtaszewski JF, Andersson L, Zierath JR. Changes in exercise-induced gene expression in 5'-AMP-activated protein kinase gamma3-null and gamma3 R225Q transgenic mice. Diabetes 2005. 54(12):3484-3489. [DOD] [CrossRef]
- Lochhead PA, Salt IP, Walker KS, Hardie DG, Sutherland C. 5-aminoimidazole-4-carboxamide riboside mimics the effects of insulin on the expression of the 2 key gluconeogenic genes PEPCK and glucose-6-phosphatase. Diabetes 2000. 49(6):896-903. [DOD] [CrossRef]
- Osler ME, Zierath JR. Adenosine 5'-monophosphate-activated protein kinase regulation of fatty acid oxidation in skeletal muscle. Endocrinology 2008. 149(3):935-941. [DOD] [CrossRef]
- Winder WW. Can patients with type 2 diabetes be treated with 5'-AMP-activated protein kinase activators? Diabetologia 2008. 51(10):1761-1764. [DOD]
- Winder WW, Holmes BF, Rubink DS, Jensen EB, Chen M, Holloszy JO. Activation of AMP-activated protein kinase increases mitochondrial enzymes in skeletal muscle. J Appl Physiol 2000. 88(6):2219-2226. [DOD]
- Winder WW, Taylor EB, Thomson DM. Role of AMP-activated protein kinase in the molecular adaptation to endurance exercise. Med Sci Sports Exerc 2006. 38(11):1945-1949. [DOD] [CrossRef]
- Marsin AS, Bertrand L, Rider MH, Deprez J, Beauloye C, Vincent MF, Van den Berghe G, Carling D, Hue L. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr Biol 2000. 10(20):1247-1255. [DOD] [CrossRef]
- Mukhtar MH, Payne VA, Arden C, Harbottle A, Khan S, Lange AJ, Agius L. Inhibition of glucokinase translocation by AMP-activated protein kinase is associated with phosphorylation of both GKRP and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase. Am J Physiol Regul Integr Comp Physiol 2008. 294(3):R766-R774. [DOD]
- Lopaschuk GD. AMP-activated protein kinase control of energy metabolism in the ischemic heart. Int J Obes (Lond) 2008. 32(Suppl 4):S29-S35. [DOD] [CrossRef]
- Russell RR 3rd, Li J, Coven DL, Pypaert M, Zechner C, Palmeri M, Giordano FJ, Mu J, Birnbaum MJ, Young LH. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J Clin Invest 2004. 114(4):495-503. [DOD]
- Wang Y, Gao E, Tao L, Lau WB, Yuan Y, Goldstein BJ, Lopez BL, Christopher TA, Tian R, Koch W, Ma XL. AMP-activated protein kinase deficiency enhances myocardial ischemia/reperfusion injury but has minimal effect on the antioxidant/antinitrative protection of adiponectin. Circulation 2009. 119(6):835-844. [DOD] [CrossRef]
- Barnes K, Ingram JC, Porras OH, Barros LF, Hudson ER, Fryer LG, Foufelle F, Carling D, Hardie DG, Baldwin SA. Activation of GLUT1 by metabolic and osmotic stress: potential involvement of AMP-activated protein kinase (AMPK). J Cell Sci 2002. 115(Pt 11):2433-2442. [DOD]
- Krook A, Wallberg-Henriksson H, Zierath JR. Sending the signal: molecular mechanisms regulating glucose uptake. Med Sci Sports Exerc 2004. 36(7):1212-1217. [DOD] [CrossRef]
- Towler MC, Hardie DG. AMP-activated protein kinase in metabolic control and insulin signaling. Circ Res 2007. 100(3):328-341. [DOD] [CrossRef]
- Beauloye C, Marsin AS, Bertrand L, Krause U, Hardie DG, Vanoverschelde JL, Hue L. Insulin antagonizes AMP-activated protein kinase activation by ischemia or anoxia in rat hearts, without affecting total adenine nucleotides. FEBS Lett 2001. 505(3):348-352. [DOD] [CrossRef]
- Gamble J, Lopaschuk GD. Insulin inhibition of 5' adenosine monophosphate-activated protein kinase in the heart results in activation of acetyl coenzyme A carboxylase and inhibition of fatty acid oxidation. Metabolism 1997. 46(11):1270-1274. [DOD] [CrossRef]
- Horman S, Vertommen D, Heath R, Neumann D, Mouton V, Woods A, Schlattner U, Wallimann T, Carling D, Hue L, Rider MH. Insulin antagonizes ischemia-induced Thr172 phosphorylation of AMP-activated protein kinase alpha-subunits in heart via hierarchical phosphorylation of Ser485/491. J Biol Chem 2006. 281(9):5335-5340. [DOD] [CrossRef]
- Holmes BF, Kurth-Kraczek EJ, Winder WW. Chronic activation of 5'-AMP-activated protein kinase increases GLUT-4, hexokinase, and glycogen in muscle. J Appl Physiol 1999. 87(5):1990-1995. [DOD]
- Jessen N, Pold R, Buhl ES, Jensen LS, Schmitz O, Lund S. Effects of AICAR and exercise on insulin-stimulated glucose uptake, signaling, and GLUT-4 content in rat muscles. J Appl Physiol 2003. 94(4):1373-1379. [DOD]
- McGee SL, Howlett KF, Starkie RL, Cameron-Smith D, Kemp BE, Hargreaves M. Exercise increases nuclear AMPK alpha2 in human skeletal muscle. Diabetes 2003. 52(4):926-928. [DOD] [CrossRef]
- McGee SL, Hargreaves M. Exercise and skeletal muscle glucose transporter 4 expression: molecular mechanisms. Clin Exp Pharmacol Physiol 2006. 33(4):395-399. [DOD] [CrossRef]
- Fisher JS, Gao J, Han DH, Holloszy JO, Nolte LA. Activation of AMP kinase enhances sensitivity of muscle glucose transport to insulin. Am J Physiol Endocrinol Metab 2002. 282(1):E18-E23. [DOD]
- Al-Khalili L, Krook A, Zierath JR, Cartee GD. Prior serum- and AICAR-induced AMPK activation in primary human myocytes does not lead to subsequent increase in insulin-stimulated glucose uptake. Am J Physiol Endocrinol Metab 2004. 287(3):E553-E557. [DOD] [CrossRef]
- Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003. 115(5):577-590. [DOD] [CrossRef]
- Mu J, Brozinick JT Jr, Valladares O, Bucan M, Birnbaum MJ. A role for AMP-activated protein kinase in contraction- and hypoxia-regulated glucose transport in skeletal muscle. Mol Cell 2001. 7(5):1085-1094. [DOD] [CrossRef]
- Funai K, Cartee GD. Inhibition of contraction-stimulated AMP-activated protein kinase inhibits contraction-stimulated increases in PAS-TBC1D1 and glucose transport without altering PAS-AS160 in rat skeletal muscle. Diabetes 2009. 58(5):1096-1104. [DOD] [CrossRef]
- Reznick RM, Zong H, Li J, Morino K, Moore IK, Yu HJ, Liu ZX, Dong J, Mustard KJ, Hawley SA, Befroy D, Pypaert M, Hardie DG, Young LH, Shulman GI. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab 2007. 5(2):151-156. [DOD] [CrossRef]
- Bavenholm PN, Pigon J, Saha AK, Ruderman NB, Efendic S. Fatty acid oxidation and the regulation of malonyl-CoA in human muscle. Diabetes 2000. 49(7):1078-1083. [DOD] [CrossRef]
- Lin J, Handschin C, Spiegelman BM. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab 2005. 1(6):361-370. [DOD] [CrossRef]
- Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009. 458(7241):1056-1060. [DOD] [CrossRef]
- Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A 2007. 104(29):12017-12022. [DOD] [CrossRef]
- Daval M, Foufelle F, Ferre P. Functions of AMP-activated protein kinase in adipose tissue. J Physiol 2006. 574(Pt 1):55-62. [DOD] [CrossRef]
- Salt IP, Connell JM, Gould GW. 5-aminoimidazole-4-carboxamide ribonucleoside (AICAR) inhibits insulin-stimulated glucose transport in 3T3-L1 adipocytes. Diabetes 2000. 49(10):1649-1656. [DOD] [CrossRef]
- Garton AJ, Campbell DG, Carling D, Hardie DG, Colbran RJ, Yeaman SJ. Phosphorylation of bovine hormone-sensitive lipase by the AMP-activated protein kinase. A possible antilipolytic mechanism. Eur J Biochem 1989. 179(1):249-254. [DOD] [CrossRef]
- Sullivan JE, Brocklehurst KJ, Marley AE, Carey F, Carling D, Beri RK. Inhibition of lipolysis and lipogenesis in isolated rat adipocytes with AICAR, a cell-permeable activator of AMP-activated protein kinase. FEBS Lett 1994. 353(1):33-36. [DOD] [CrossRef]
- Daval M, Diot-Dupuy F, Bazin R, Hainault I, Viollet B, Vaulont S, Hajduch E, Ferre P, Foufelle F. Anti-lipolytic action of AMP-activated protein kinase in rodent adipocytes. J Biol Chem 2005. 280(26):25250-25257. [DOD] [CrossRef]
- Villena JA, Viollet B, Andreelli F, Kahn A, Vaulont S, Sul HS. Induced adiposity and adipocyte hypertrophy in mice lacking the AMP-activated protein kinase-alpha2 subunit. Diabetes 2004. 53(9):2242-2249. [DOD] [CrossRef]
- Qi L, Saberi M, Zmuda E, Wang Y, Altarejos J, Zhang X, Dentin R, Hedrick S, Bandyopadhyay G, Hai T, Olefsky J, Montminy M. Adipocyte CREB promotes insulin resistance in obesity. Cell Metab 2009. 9(3):277-286. [DOD]
- Andreelli F, Foretz M, Knauf C, Cani PD, Perrin C, Iglesias MA, Pillot B, Bado A, Tronche F, Mithieux G, Vaulont S, Burcelin R, Viollet B. Liver adenosine monophosphate-activated kinase-alpha2 catalytic subunit is a key target for the control of hepatic glucose production by adiponectin and leptin but not insulin. Endocrinology 2006. 147(5):2432-2441. [DOD] [CrossRef]
- Foretz M, Ancellin N, Andreelli F, Saintillan Y, Grondin P, Kahn A, Thorens B, Vaulont S, Viollet B. Short-term overexpression of a constitutively active form of AMP-activated protein kinase in the liver leads to mild hypoglycemia and fatty liver. Diabetes 2005. 54(5):1331-1339. [DOD] [CrossRef]
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 2001. 108(8):1167-1174. [DOD]
- Ben Djoudi Ouadda A, Levy E, Ziv E, Lalonde G, Sane AT, Delvin E, Elchebly M. Increased hepatic lipogenesis in insulin resistance and type 2 diabetes is associated to AMPK signaling pathway upregulation in Psammomys obesus. Biosci Rep 2008. 29:283-292. [DOD]
- Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med 2002. 8(11):1288-1295. [DOD] [CrossRef]
- Viollet B, Mounier R, Leclerc J, Yazigi A, Foretz M, Andreelli F. Targeting AMP-activated protein kinase as a novel therapeutic approach for the treatment of metabolic disorders. Diabetes Metab 2007. 33(6):395-402. [DOD] [CrossRef]
- Koo SH, Flechner L, Qi L, Zhang X, Screaton RA, Jeffries S, Hedrick S, Xu W, Boussouar F, Brindle P, Takemori H, Montminy M. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature 2005. 437(7062):1109-1111. [DOD] [CrossRef]
- Velasco G, Geelen MJ, Guzman M. Control of hepatic fatty acid oxidation by 5'-AMP-activated protein kinase involves a malonyl-CoA-dependent and a malonyl-CoA-independent mechanism. Arch Biochem Biophys 1997. 337(2):169-175. [DOD] [CrossRef]
- Kim KH. Regulation of mammalian acetyl-coenzyme A carboxylase. Annu Rev Nutr 1997. 17:77-99. [DOD] [CrossRef]
- Rutter GA, Leclerc I. The AMP-regulated kinase family: enigmatic targets for diabetes therapy. Mol Cell Endocrinol 2009. 297(1-2):41-49. [DOD] [CrossRef]
- Lupi R, Marchetti P, Giannarelli R, Coppelli A, Tellini C, Del Guerra S, Lorenzetti M, Carmellini M, Mosca F, Navalesi R. Effects of glibenclamide and metformin (alone or in combination) on insulin release from isolated human pancreatic islets. Acta Diabetol 1997. 34(1):46-48. [DOD] [CrossRef]
- Leclerc I, Woltersdorf WW, da Silva Xavier G, Rowe RL, Cross SE, Korbutt GS, Rajotte RV, Smith R, Rutter GA. Metformin, but not leptin, regulates AMP-activated protein kinase in pancreatic islets: impact on glucose-stimulated insulin secretion. Am J Physiol Endocrinol Metab 2004. 286(6):E1023-E1031. [DOD] [CrossRef]
- da Silva Xavier G, Leclerc I, Salt IP, Doiron B, Hardie DG, Kahn A, Rutter GA. Role of AMP-activated protein kinase in the regulation by glucose of islet beta cell gene expression. Proc Natl Acad Sci U S A 2000. 97(8):4023-4028. [DOD] [CrossRef]
- Zhang J, Xie Z, Dong Y, Wang S, Liu C, Zou MH. Identification of nitric oxide as an endogenous activator of the AMP-activated protein kinase in vascular endothelial cells. J Biol Chem 2008. 283(41):27452-27461. [DOD] [CrossRef]
- Procopio C, Andreozzi F, Laratta E, Cassese A, Beguinot F, Arturi F, Hribal ML, Perticone F, Sesti G. Leptin-stimulated endothelial nitric-oxide synthase via an AMPK/Akt signalling pathway is attenuated by interaction with C reactive protein. Endocrinology 2009. In press. [DOD]
- Wang W, Fan J, Yang X, Furer-Galban S, Lopez de Silanes I, von Kobbe C, Guo J, Georas SN, Foufelle F, Hardie DG, Carling D, Gorospe M. AMP-activated kinase regulates cytoplasmic HuR. Mol Cell Biol 2002. 22(10):3425-3436. [DOD] [CrossRef]
- Horman S, Browne G, Krause U, Patel J, Vertommen D, Bertrand L, Lavoinne A, Hue L, Proud C, Rider M. Activation of AMP-activated protein kinase leads to the phosphorylation of elongation factor 2 and an inhibition of protein synthesis. Curr Biol 2002. 12(16):1419-1423. [DOD] [CrossRef]
- Bowker SL, Majumdar SR, Veugelers P, Johnson JA. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care 2006. 29(2):254-258. [DOD] [CrossRef]
- Kuhajda FP. AMP-activated protein kinase and human cancer: cancer metabolism revisited. Int J Obes (Lond) 2008. 32(Suppl 4):S36-S41. [DOD] [CrossRef]
- Neumeier M, Weigert J, Schaffler A, Wehrwein G, Muller-Ladner U, Scholmerich J, Wrede C, Buechler C. Different effects of adiponectin isoforms in human monocytic cells. J Leukoc Biol 2006. 79(4):803-808. [DOD] [CrossRef]
- Guest CB, Chakour KS, Freund GG. Macropinocytosis is decreased in diabetic mouse macrophages and is regulated by AMPK. BMC Immunol 2008. 9:42. [DOD] [CrossRef]
- Minokoshi Y, Alquier T, Furukawa N, Kim YB, Lee A, Xue B, Mu J, Foufelle F, Ferre P, Birnbaum MJ, Stuck BJ, Kahn BB. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 2004. 428(6982):569-574. [DOD] [CrossRef]
- Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, Cantley LC. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 2005. 310(5754):1642-1646. [DOD] [CrossRef]
- Kola B, Boscaro M, Rutter GA, Grossman AB, Korbonits M. Expanding role of AMPK in endocrinology. Trends Endocrinol Metab 2006. 17(5):205-215. [DOD] [CrossRef]
- Kongsuphol P, Cassidy D, Hieke B, Treharne KJ, Schreiber R, Mehta A, Kunzelmann K. Mechanistic insight into control of CFTR by AMPK. J Biol Chem 2009. 284(9):5645-5653. [DOD] [CrossRef]
- Bailey CJ, Turner RC. Metformin. N Engl J Med 1996. 334(9):574-579. [DOD] [CrossRef]
- Hawley SA, Gadalla AE, Olsen GS, Hardie DG. The antidiabetic drug metformin activates the AMP-activated protein kinase cascade via an adenine nucleotide-independent mechanism. Diabetes 2002. 51(8):2420-2425. [DOD]
- Sakamoto K, Goransson O, Hardie DG, Alessi DR. Activity of LKB1 and AMPK-related kinases in skeletal muscle: effects of contraction, phenformin, and AICAR. Am J Physiol Endocrinol Metab 2004. 287(2):E310-E317. [DOD] [CrossRef]
- Chen Y, Zhou K, Wang R, Liu Y, Kwak YD, Ma T, Thompson RC, Zhao Y, Smith L, Gasparini L, Luo Z, Xu H, Liao FF. Antidiabetic drug metformin (GlucophageR) increases biogenesis of Alzheimer's amyloid peptides via up-regulating BACE1 transcription. Proc Natl Acad Sci U S A 2009. 106(10):3907-3912. [DOD] [CrossRef]
- Schimmack G, Defronzo RA, Musi N. AMP-activated protein kinase: Role in metabolism and therapeutic implications. Diabetes Obes Metab 2006. 8(6):591-602. [DOD] [CrossRef]
- Bays H, Mandarino L, DeFronzo RA. Role of the adipocyte, free fatty acids, and ectopic fat in pathogenesis of type 2 diabetes mellitus: peroxisomal proliferator-activated receptor agonists provide a rational therapeutic approach. J Clin Endocrinol Metab 2004. 89(2):463-478. [DOD] [CrossRef]
- Wu X, Motoshima H, Mahadev K, Stalker TJ, Scalia R, Goldstein BJ. Involvement of AMP-activated protein kinase in glucose uptake stimulated by the globular domain of adiponectin in primary rat adipocytes. Diabetes 2003. 52(6):1355-1363. [DOD] [CrossRef]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 2003. 34(3):267-273. [DOD] [CrossRef]
- Bogacka I, Xie H, Bray GA, Smith SR. Pioglitazone induces mitochondrial biogenesis in human subcutaneous adipose tissue in vivo. Diabetes 2005. 54(5):1392-1399. [DOD] [CrossRef]
- Coletta DK, Sriwijitkamol A, Wajcberg E, Tantiwong P, Li M, Prentki M, Madiraju M, Jenkinson CP, Cersosimo E, Musi N, Defronzo RA. Pioglitazone stimulates AMP-activated protein kinase signalling and increases the expression of genes involved in adiponectin signalling, mitochondrial function and fat oxidation in human skeletal muscle in vivo: a randomised trial. Diabetologia 2009. 52(4):723-732. [DOD] [CrossRef]
- Fryer LG, Parbu-Patel A, Carling D. The Anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J Biol Chem 2002. 277(28):25226-25232. [DOD] [CrossRef]
- Zhang F, Dey D, Branstrom R, Forsberg L, Lu M, Zhang Q, Sjoholm A. BLX-1002, a novel thiazolidinedione with no PPAR affinity, stimulates AMP-activated protein kinase activity, raises cytosolic Ca2+, and enhances glucose-stimulated insulin secretion in a PI3K-dependent manner. Am J Physiol Cell Physiol 2009. 296(2):C346-C354. [DOD] [CrossRef]
- Winder WW, Hardie DG. AMP-activated protein kinase, a metabolic master switch: possible roles in type 2 diabetes. Am J Physiol 1999. 277(1 Pt 1):E1-E10. [DOD]
- Gruzman A, Shamni O, Ben Yakir M, Sandovski D, Elgart A, Alpert E, Cohen G, Hoffman A, Katzhendler Y, Cerasi E, Sasson S. Novel D-xylose derivatives stimulate muscle glucose uptake by activating AMP-activated protein kinase alpha. J Med Chem 2008. 51(24):8096-8108. [DOD] [CrossRef]
- Brusq JM, Ancellin N, Grondin P, Guillard R, Martin S, Saintillan Y, Issandou M. Inhibition of lipid synthesis through activation of AMP kinase: an additional mechanism for the hypolipidemic effects of berberine. J Lipid Res 2006. 47(6):1281-1288. [DOD] [CrossRef]
- Turner N, Li JY, Gosby A, To SW, Cheng Z, Miyoshi H, Taketo MM, Cooney GJ, Kraegen EW, James DE, Hu LH, Li J, Ye JM. Berberine and its more biologically available derivative, dihydroberberine, inhibit mitochondrial respiratory complex I: a mechanism for the action of berberine to activate AMP-activated protein kinase and improve insulin action. Diabetes 2008. 57(5):1414-1418. [DOD] [CrossRef]
- Lee YS, Kim WS, Kim KH, Yoon MJ, Cho HJ, Shen Y, Ye JM, Lee CH, Oh WK, Kim CT, et al. Berberine, a natural plant product, activates AMP-activated protein kinase with beneficial metabolic effects in diabetic and insulin-resistant states. Diabetes 2006. 55(8):2256-2264. [DOD] [CrossRef]
- Kosaka T, Okuyama R, Sun W, Ogata T, Harada J, Araki K, Izumi M, Yoshida T, Okuno A, Fujiwara T, Ohsumi J, Ichikawa K. Identification of molecular target of AMP-activated protein kinase activator by affinity purification and mass spectrometry. Anal Chem 2005. 77(7):2050-2055. [DOD] [CrossRef]
- Suzuki K, Uchida K, Nakanishi N, Hattori Y. Cilostazol activates AMP-activated protein kinase and restores endothelial function in diabetes. Am J Hypertens 2008. 21(4):451-457. [DOD] [CrossRef]
- Ma J, Zheng M, Shen Y, Wang L, Zhu Y, Zhang X. Effects of cilostazol on thrombospondin-1 (TSP-1) expression changes in the myocardium of diabetic rats. J Int Med Res 2008. 36(4):800-809. [DOD]
- Park SY, Shin HK, Lee JH, Kim CD, Lee WS, Rhim BY, Hong KW. Cilostazol ameliorates metabolic abnormalities with suppression of proinflammatory markers in a db/db mouse model of type 2 diabetes via activation of peroxisome proliferator-activated receptor gamma transcription. J Pharmacol Exp Ther 2009. 329(2):571-579. [DOD] [CrossRef]
- O'Donnell ME, Badger SA, Sharif MA, Makar RR, Young IS, Lee B, Soong CV. The vascular and biochemical effects of cilostazol in diabetic patients with peripheral arterial disease. Vasc Endovascular Surg 2009. 43(2):132-143. [DOD] [CrossRef]
- Zhou G, Sebhat IK, Zhang BB. AMPK activators--potential therapeutics for metabolic and other diseases. Acta Physiol (Oxf) 2009. 196(1):175-190. [DOD] [CrossRef]
- Lee NK, Lee JW, Lee S, Im GJ, Han HY, Kim TK, et al. Quinazoline derivatives for the treatment and prevention of diabetes and obesity. Patent no. WO/2006/071095. Publ. date 08.06.2006. Available online at http://www.wipo.int/pctdb/en/wo.jsp?wo=2006071095. [DOD]
- Singh R, Yu J, Hong H, Thota S, Xu X. N-substituted-heterocycloalkyloxybenzamide compounds and methods of use. Patent no. WO/2008/083124. Publ. date 10.07.2008. Available online at http://www.wipo.int/pctdb/en/wo.jsp?WO=2008083124. [DOD]
- Cederroth CR, Vinciguerra M, Gjinovci A, Kuhne F, Klein M, Cederroth M, Caille D, Suter M, Neumann D, James RW, et al. Dietary phytoestrogens activate AMP-activated protein kinase with improvement in lipid and glucose metabolism. Diabetes 2008. 57(5):1176-1185. [DOD] [CrossRef]
- Ahn J, Lee H, Kim S, Park J, Ha T. The anti-obesity effect of quercetin is mediated by the AMPK and MAPK signaling pathways. Biochem Biophys Res Commun 2008. 373(4):545-549. [DOD] [CrossRef]
- Collins QF, Liu HY, Pi J, Liu Z, Quon MJ, Cao W. Epigallocatechin-3-gallate (EGCG), a green tea polyphenol, suppresses hepatic gluconeogenesis through 5'-AMP-activated protein kinase. J Biol Chem 2007. 282(41):30143-30149. [DOD] [CrossRef]
- Kusirisin W, Srichairatanakool S, Lerttrakarnnon P, Lailerd N, Suttajit M, Jaikang C, Chaiyasut C. Antioxidative activity, polyphenolic content and anti-glycation effect of some Thai medicinal plants traditionally used in diabetic patients. Med Chem 2009. 5(2):139-147. [DOD] [CrossRef]
- Liu J, Sun H, Duan W, Mu D, Zhang L. Maslinic acid reduces blood glucose in KK-Ay mice. Biol Pharm Bull 2007. 30(11):2075-2078. [DOD] [CrossRef]
- Tan MJ, Ye JM, Turner N, Hohnen-Behrens C, Ke CQ, Tang CP, Chen T, Weiss HC, Gesing ER, Rowland A, James DE, Ye Y. Antidiabetic activities of triterpenoids isolated from bitter melon associated with activation of the AMPK pathway. Chem Biol 2008. 15(3):263-273. [DOD] [CrossRef]
- Park MW, Ha J, Chung SH. 20(S)-ginsenoside Rg3 enhances glucose-stimulated insulin secretion and activates AMPK. Biol Pharm Bull 2008. 31(4):748-751. [DOD] [CrossRef]
- Hwang JT, Lee MS, Kim HJ, Sung MJ, Kim HY, Kim MS, Kwon DY. Antiobesity effect of ginsenoside Rg3 involves the AMPK and PPAR-gamma signal pathways. Phytother Res 2009. 23(2):262-266. [DOD] [CrossRef]
- Hwang SL, Yang BK, Lee JY, Kim JH, Kim BD, Suh KH, Kim DY, Kim MS, Song H, Park BS, Huh TL. Isodihydrocapsiate stimulates plasma glucose uptake by activation of AMP-activated protein kinase. Biochem Biophys Res Commun 2008. 371(2):289-293. [DOD] [CrossRef]
- Li HB, Ge YK, Zheng XX, Zhang L. Salidroside stimulated glucose uptake in skeletal muscle cells by activating AMP-activated protein kinase. Eur J Pharmacol 2008. 588(2-3):165-169. [DOD] [CrossRef]
- Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006. 444(7117):337-342. [DOD] [CrossRef]
- Pettit GR, Singh SB, Hamel E, Lin CM, Alberts DS, Garcia-Kendall D. Isolation and structure of the strong cell growth and tubulin inhibitor combretastatin A-4. Experientia 1989. 45(2):209-211. [DOD]
- Zhang F, Sun C, Wu J, He C, Ge X, Huang W, Zou Y, Chen X, Qi W, Zhai Q. Combretastatin A-4 activates AMP-activated protein kinase and improves glucose metabolism in db/db mice. Pharmacol Res 2008. 57(4):318-323. [DOD] [CrossRef]
- Lopez-Lazaro M. Anticancer and carcinogenic properties of curcumin: considerations for its clinical development as a cancer chemopreventive and chemotherapeutic agent. Mol Nutr Food Res 2008. 52(Suppl 1):S103-S127. [DOD]
- Pan W, Yang H, Cao C, Song X, Wallin B, Kivlin R, Lu S, Hu G, Di W, Wan Y. AMPK mediates curcumin-induced cell death in CaOV3 ovarian cancer cells. Oncol Rep 2008. 20(6):1553-1559. [DOD]
- Kanitkar M, Gokhale K, Galande S, Bhonde RR. Novel role of curcumin in the prevention of cytokine-induced islet death in vitro and diabetogenesis in vivo. Br J Pharmacol 2008. 155(5):702-713. [DOD] [CrossRef]
- Fujiwara H, Hosokawa M, Zhou X, Fujimoto S, Fukuda K, Toyoda K, Nishi Y, Fujita Y, Yamada K, Yamada Y, Seino Y, Inagaki N. Curcumin inhibits glucose production in isolated mice hepatocytes. Diabetes Res Clin Pract 2008. 80(2):185-191. [DOD] [CrossRef]
- Cool B, Zinker B, Chiou W, Kifle L, Cao N, Perham M, Dickinson R, Adler A, Gagne G, Iyengar R, et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab 2006. 3(6):403-416. [DOD] [CrossRef]
- Goransson O, McBride A, Hawley SA, Ross FA, Shpiro N, Foretz M, Viollet B, Hardie DG, Sakamoto K. Mechanism of action of A-769662, a valuable tool for activation of AMP-activated protein kinase. J Biol Chem 2007. 282(45):32549-32560. [DOD] [CrossRef]
- Use of thienopyridone derivatives as AMPK activators and pharmaceutical compositions containing them. European Patent Application no. EP1754483. Available online at http://www.freepatentsonline.com/EP1754483A1.html. [DOD]
- Vincent MF, Erion MD, Gruber HE, Van den Berghe G. Hypoglycaemic effect of AICAriboside in mice. Diabetologia 1996. 39(10):1148-1155. [DOD] [CrossRef]
- Bergeron R, Russell RR 3rd, Young LH, Ren JM, Marcucci M, Lee A, Shulman GI. Effect of AMPK activation on muscle glucose metabolism in conscious rats. Am J Physiol 1999. 276(5 Pt 1):E938-E944. [DOD]
- Babraj JA, Mustard K, Sutherland C, Towler MC, Chen S, Smith K, Green K, Leese G, Hardie DG, Rennie MJ, Cuthbertson DJ. Blunting of AICAR-induced human skeletal muscle glucose uptake in type 2 diabetes is dependent on age rather than diabetic status. Am J Physiol Endocrinol Metab 2009. 296(5):E1042-E1048. [DOD] [CrossRef]
- Xie Z, Zhang J, Wu J, Viollet B, Zou MH. Upregulation of mitochondrial uncoupling protein-2 by the AMP-activated protein kinase in endothelial cells attenuates oxidative stress in diabetes. Diabetes 2008. 57(12):3222-3230. [DOD] [CrossRef]
- Lihn AS, Pedersen SB, Lund S, Richelsen B. The anti-diabetic AMPK activator AICAR reduces IL-6 and IL-8 in human adipose tissue and skeletal muscle cells. Mol Cell Endocrinol 2008. 292(1-2):36-41. [DOD] [CrossRef]
- Goodyear LJ. The exercise pill--too good to be true? N Engl J Med 2008. 359(17):1842-1844. [DOD]
- Fabianowska-Majewska K, Duley JA, Simmonds HA. Effects of novel anti-viral adenosine analogues on the activity of S-adenosylhomocysteine hydrolase from human liver. Biochem Pharmacol 1994. 48(5):897-903. [DOD] [CrossRef]
- Charton J, Girault-Mizzi S, Debreu-Fontaine MA, Foufelle F, Hainault I, Bizot-Espiard JG, Caignard DH, Sergheraert C. Synthesis and biological evaluation of benzimidazole derivatives as potent AMP-activated protein kinase activators. Bioorg Med Chem 2006. 14(13):4490-4518. [DOD] [CrossRef]
- Moinet G, Marais D, Hallakou-Bozec S, Charon C. Use of AMPK-activating imidazole derivatives, preparation process and pharmaceutical compositions comprising them. Patent no. WO/2008/006432. Publ. date 17.01.2008. Available online at http://www.wipo.int/pctdb/en/wo.jsp?WO=2008006432. [DOD]
- Rault S, Lancelot JC, Kopp M, Caignard DH, Pfeiffer B, Renard P, Bizot Espiard JG. New imidazopyridine derivatives are AMP activated protein kinase activators, useful in the treatment of diabetes, hypercholesterolemia, hyperlipidemia, obesity, and diabetic complications in cardiovascular system. Patent no. FR2846656. Publ. date 07.05.2007. Available online at http://v3.espacenet.com/publicationDetails/biblio;jsessionid=CAEAE2C99617116CCB3C15CD0E4B35C3.espacenet_levelx_prod_7?CC=FR&NR=2846656A1&KC=A1&FT=D&date=20040507&DB=&locale=. [DOD]
- Wang YQ, Dong Y, Yao MH. Chromium picolinate inhibits resistin secretion in insulin-resistant 3T3-L1 adipocytes through the activation of AMP activated protein kinase. Clin Exp Pharmacol Physiol 2009. In press. [DOD]
- Arner P. Resistin: yet another adipokine tells us that men are not mice. Diabetologia 2005. 48(11):2203-2205. [DOD] [CrossRef]
- Park KG, Min AK, Koh EH, Kim HS, Kim MO, Park HS, Kim YD, Yoon TS, Jang BK, Hwang JS, et al. Alpha-lipoic acid decreases hepatic lipogenesis through adenosine monophosphate-activated protein kinase (AMPK)-dependent and AMPK-independent pathways. Hepatology 2008. 48(5):1477-1486. [DOD] [CrossRef]
- Shen QW, Zhu MJ, Tong J, Ren J, Du M. Ca2+/calmodulin-dependent protein kinase kinase is involved in AMP-activated protein kinase activation by alpha-lipoic acid in C2C12 myotubes. Am J Physiol Cell Physiol 2007. 293(4):C1395-C1403. [DOD] [CrossRef]
- Yoon H, Oh YT, Lee JY, Choi JH, Lee JH, Baik HH, Kim SS, Choe W, Yoon KS, Ha J, Kang I. Activation of AMP-activated protein kinase by kainic acid mediates brain-derived neurotrophic factor expression through a NF-kappaB dependent mechanism in C6 glioma cells. Biochem Biophys Res Commun 2008. 371(3):495-500. [DOD] [CrossRef]
- Kola B, Hubina E, Tucci SA, Kirkham TC, Garcia EA, Mitchell SE, Williams LM, Hawley SA, Hardie DG, Grossman AB, Korbonits M. Cannabinoids and ghrelin have both central and peripheral metabolic and cardiac effects via AMP-activated protein kinase. J Biol Chem 2005. 280(26):25196-25201. [DOD] [CrossRef]
- Za'tara G, Bar-Tana J, Kalderon B, Suter M, Morad E, Samovski D, Neumann D, Hertz R. AMPK activation by long chain fatty acyl analogs. Biochem Pharmacol 2008. 76(10):1263-1275. [DOD] [CrossRef]
- Gao Z, Yin J, Zhang J, Ward RE, Martin RJ, Lefevre M, Cefalu WT, Ye J. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 2009. In press. [DOD]
- Toyoda T, Hayashi T, Miyamoto L, Yonemitsu S, Nakano M, Tanaka S, Ebihara K, Masuzaki H, Hosoda K, Inoue G, Otaka A, Sato K, Fushiki T, Nakao K. Possible involvement of the alpha1 isoform of 5'AMP-activated protein kinase in oxidative stress-stimulated glucose transport in skeletal muscle. Am J Physiol Endocrinol Metab 2004. 287(1):E166-E173. [DOD] [CrossRef]
- Zou MH, Hou XY, Shi CM, Kirkpatick S, Liu F, Goldman MH, Cohen RA. Activation of 5'-AMP-activated kinase is mediated through c-Src and phosphoinositide 3-kinase activity during hypoxia-reoxygenation of bovine aortic endothelial cells. Role of peroxynitrite. J Biol Chem 2003. 278(36):34003-34010. [DOD] [CrossRef]
- Kusakabe T, Tanioka H, Ebihara K, Hirata M, Miyamoto L, Miyanaga F, Hige H, Aotani D, Fujisawa T, Masuzaki H, Hosoda K, Nakao K. Beneficial effects of leptin on glycaemic and lipid control in a mouse model of type 2 diabetes with increased adiposity induced by streptozotocin and a high-fat diet. Diabetologia 2009. 52(4):675-683. [DOD] [CrossRef]
- Tanaka T, Hidaka S, Masuzaki H, Yasue S, Minokoshi Y, Ebihara K, Chusho H, Ogawa Y, Toyoda T, Sato K, et al. Skeletal muscle AMP-activated protein kinase phosphorylation parallels metabolic phenotype in leptin transgenic mice under dietary modification. Diabetes 2005. 54(8):2365-2374. [DOD] [CrossRef]
- Andersson U, Filipsson K, Abbott CR, Woods A, Smith K, Bloom SR, Carling D, Small CJ. AMP-activated protein kinase plays a role in the control of food intake. J Biol Chem 2004. 279(13):12005-12008. [DOD] [CrossRef]
- Vestergaard ET, Gormsen LC, Jessen N, Lund S, Hansen TK, Moller N, Jorgensen JO. Ghrelin infusion in humans induces acute insulin resistance and lipolysis independent of growth hormone signaling. Diabetes 2008. 57(12):3205-3210. [DOD] [CrossRef]
- Anderson KA, Ribar TJ, Lin F, Noeldner PK, Green MF, Muehlbauer MJ, Witters LA, Kemp BE, Means AR. Hypothalamic CaMKK2 contributes to the regulation of energy balance. Cell Metab 2008. 7(5):377-388. [DOD] [CrossRef]
- Carey AL, Steinberg GR, Macaulay SL, Thomas WG, Holmes AG, Ramm G, Prelovsek O, Hohnen-Behrens C, Watt MJ, James DE, Kemp BE, Pedersen BK, Febbraio MA. Interleukin-6 increases insulin-stimulated glucose disposal in humans and glucose uptake and fatty acid oxidation in vitro via AMP-activated protein kinase. Diabetes 2006. 55(10):2688-2697. [DOD] [CrossRef]
- Fluckiger-Isler RE, Walter P. Stimulation of rat liver glycogen synthesis by the adenosine kinase inhibitor 5-iodotubercidin. Biochem J 1993. 292(Pt 1):85-91. [DOD]
- Fox T, Coll JT, Xie X, Ford PJ, Germann UA, Porter MD, Pazhanisamy S, Fleming MA, Galullo V, Su MS, Wilson KP. A single amino acid substitution makes ERK2 susceptible to pyridinyl imidazole inhibitors of p38 MAP kinase. Protein Sci 1998. 7(11):2249-2255. [DOD] [CrossRef]
- Garcia-Villafranca J, Castro J. Effects of 5-iodotubercidin on hepatic fatty acid metabolism mediated by the inhibition of acetyl-CoA carboxylase. Biochem Pharmacol 2002. 63(11):1997-2000. [DOD] [CrossRef]
- Massillon D, Stalmans W, van de Werve G, Bollen M. Identification of the glycogenic compound 5-iodotubercidin as a general protein kinase inhibitor. Biochem J 1994. 299(Pt 1):123-128. [DOD]
- Russell RR 3rd, Bergeron R, Shulman GI, Young LH. Translocation of myocardial GLUT-4 and increased glucose uptake through activation of AMPK by AICAR. Am J Physiol 1999. 277(2 Pt 2):H643-H649. [DOD]
- Musi N, Hayashi T, Fujii N, Hirshman MF, Witters LA, Goodyear LJ. AMP-activated protein kinase activity and glucose uptake in rat skeletal muscle. Am J Physiol Endocrinol Metab 2001. 280(5):E677-E684. [DOD]
- Hardie DG, Carling D. The AMP-activated protein kinase--fuel gauge of the mammalian cell? Eur J Biochem 1997. 246(2):259-273. [DOD]
This article has been cited by other articles:

|
Molecular mechanisms for anti-aging by natural dietary compounds
Pan MH, Lai CS, Tsai ML, Wu JC, Ho CT
Mol Nutr Food Res 2012. 56(1):88-115
|
|
|
Peptide scaffolds: flexible molecular structures with diverse therapeutic potentials
Deshmukh R, Purohit HJ
Int J Peptide Res Ther 2012. In press
|
|

|
AMPK protects proximal tubular cells from stress-induced apoptosis by an ATP-independent mechanism: potential role of Akt activation
Lieberthal W, Zhang L, Patel VA, Levine JS
Am J Physiol Renal Physiol 2011. 301(6):F1177-F1192
|
|

|
Computer-aided drug design for AMP-activated protein kinase activators
Wang Z, Huo J, Sun L, Wang Y, Jin H, Yu H, Zhang L, Zhou L
Curr Comput Aided Drug Des 2011. 7(3):214-227
|
|

|
Effect of chromium enriched fermentation product of barley and brewer's yeast and its combination with rosiglitazone on experimentally induced hyperglycaemia in mice
Cekic V, Vasovic V, Jakovljevic V, Lalosevic D, Capo I, Mikov M, Sabo A
Srp Arh Celok Lek 2011. 139(9-10):610-618
|
|

|
Antihyperglycaemic activity of 2,4:3,5-dibenzylidene-D-xylose-dithioacetal in diabetic mice
Gruzman A, Elgart A, Viskind O, Billauer H, Dotan S, Cohen G, Mishani E, Hoffman A, Cerasi E, Sasson S
J Cell Mol Med 2011. In press
|
|
|
Nutrigenomics of neuradaptogen amino-acid-therapy and neurometabolic optimizers: overcoming carbohydrate bingeing and overeating through neurometabolic mechanisms
Blum K, Bagchi D, Bowirrat A, Downs BW, Waite RL, Giordano J, Morse S, Madigan M, Downs JM, Braverman ER, Polanin M, Barh D, Fornari F, Simpatico T
Funct Food Health Dis 2011. 9:310-378
|
|

|
Key regulators of mitochondrial biogenesis are increased in kidneys of growth hormone receptor knockout (GHRKO) mice
Gesing A, Bartke A, Wang F, Karbownik-Lewinska M, Masternak MM
Cell Biochem Funct 2011. 29(6):459-467
|
|
|
Targeting the AMP-regulated kinase family to treat diabetes: a research update
Sun G, Rutter GA
Diabetes Manag 2011. 1(3):333-347
|
|

|
Insulin resistance and other metabolic risk factors in the pathogenesis of hepatocellular carcinoma
Siddique A, Kowdley KV
Clin Liver Dis 2011. 15(2):281-296
|
|

|
Curcumin protects hepatic stellate cells against leptin-induced activation in vitro by accumulating intracellular lipids
Tang Y, Chen A
Endocrinology 2010. 151(9):4168-4177
|
|

|
Binding of cordycepin monophosphate to AMP-activated protein kinase and its effect on AMP-activated protein kinase activation
Wang Z, Wang X, Qu K, Zhu P, Guo N, Zhang R, Abliz Z, Yu H, Zhu H
Chem Biol Drug Des 2010. 76(4):340-344
|
|

|
AMP-Activated protein kinase as a target for preconditioning in transplantation medicine
Bouma HR, Ketelaar ME, Yard BA, Ploeg RJ, Henning RH
Transplantation 2010. 90(4):353-358
|
|

|
Does LKB1 Mediate Activation of Hepatic AMP-Protein Kinase (AMPK) and Sirtuin1 (SIRT1) After Roux-en-Y Gastric Bypass in Obese Rats?
Peng Y, Rideout DA, Rakita SS, Gower WR Jr, You M, Murr MM
J Gastrointest Surg 2010. 14(2):221-228
|
|

|
Curcumin activates AMPK and suppresses gluconeogenic gene expression in hepatoma cells
Kim T, Davis J, Zhang AJ, He X, Mathews ST
Biochem Biophys Res Commun 2009. 388(2):377-382
|
|
|


)
)
)
)
)
)
)
)
)
)
)

