Chapter I. Pathogenesis
| Rev Diabet Stud,
2012,
9(4):224-235 |
DOI 10.1900/RDS.2012.9.224 |
Novel Biomarkers in Type 1 Diabetes
Yulan Jin, Jin-Xiong She
Center for Biotechnology and Genomic Medicine and Department of Pathology, Medical College of Georgia, Georgia Regents University, Augusta, GA, USA
Address correspondence to: Jin-Xiong She, Center for Biotechnology and Genomic Medicine, Medical College of Georgia, Georgia Regents University,1120 15th Street, Augusta, GA 30912, USA, e-mail: jshe@gru.edu
Manuscript submitted December 16, 2012; resubmitted December 21, 2012; accepted January 8, 2013.
Keywords: type 1 diabetes, biomarker, prediction, pathogenesis, risk, prevention, antibody, INS, CTLA4, PTPN22, IL2RA
Abstract
Biomarkers are useful tools for research into type 1 diabetes (T1D) for a number of purposes, including elucidation of disease pathogenesis, risk prediction, and therapeutic monitoring. Susceptibility genes and islet autoantibodies are currently the most useful biomarkers for T1D risk prediction. However, these markers do not fully meet the needs of scientists and physicians for several reasons. First, improvement of the specificity and sensitivity is still desirable to achieve better positive predictive values. Second, autoantibodies appear relatively late in the disease process, thus limiting their value in early disease prediction. Third, the currently available biomarkers are not useful for assessing therapeutic outcomes because some are not involved in the disease process (autoantibodies) and others do not change during disease progression (susceptibility genes). Therefore, considerable effort has been devoted to the discovery of novel T1D biomarkers in the last three decades. The advent of high-throughput technologies for genetic, transcriptomic, and proteomic studies has allowed genome-wide examinations of genetic polymorphisms, global gene changes, and protein expression changes in T1D patients and prediabetic subjects. These large-scale studies resulted in the discovery of a large number of susceptibility genes and changes in gene and protein expression. While these studies have provided a number of novel biomarker candidates, their clinical benefits remain to be evaluated in prospective studies, and no new "star biomarker" has been identified until now. Previous studies suggest that significant improvements in study design and analytical methodologies have to be made to identify clinically relevant biomarkers. In this review, we discuss progress, opportunities, challenges, and future directions in the development of T1D biomarkers, mainly by focusing on the genetic, transcriptomic, and proteomic aspects.
Abbreviations: 2D – two-dimensional; cDNA – complementary DNA; CRP – C-reactive protein; CTLA4 – cytotoxic T lymphocyte antigen 4; GADA – glutamic acid decarboxylase autoantibodies; GM-CSF – granulocyte-macrophage colony-stimulating factor; GNLY – granulysin; GWAS – genome-wide association study; GZMB – granzyme B; HLA – human leukocyte antigen; IAA – insulin autoantibodies; IA-2A – insulinoma 2-associated autoantibodies; ICA – islet cell cytoplasmic autoantibodies; IFIH1 – interferon-induced helicase c domain-containing protein 1; IFN-γ – interferon gamma; IL – interleukin; IL2RA – interleukin 2 receptor alpha; IP-10 – interferon-inducible protein of 10 kDa (CXCL10); MCP-1 – monocyte chemoattractant protein-1; MS – mass spectrometry; NFκB – nuclear factor 'kappa-light-chain-enhancer' of activated B-cells; NGS – next-generation DNA sequencing; NK – natural killer; NO – nitric oxide; PBMC – peripheral blood mononuclear cell; PTPN22 – protein tyrosine phosphatase, non-receptor type 22; RNA – ribonucleic acid; RT-PCR – real-time polymerase chain reaction; SAA – serum amyloid protein A; SELL – selectin L; SLE – systemic lupus erythematosus; SNP – single nucleotide polymorphism; T1D – type 1 diabetes; TGF – transforming growth factor; TNFα – tumor necrosis factor alpha; ZnT8A – zinc transporter 8 antibody
1. Introduction
Type 1 diabetes (T1D) is an autoimmune disease primarily starting in childhood. It results from the destruction of insulin-producing β-cells of the pancreas [1]. Although exogenous insulin can help to maintain the level of blood glucose, there is no cure for this disease, and long-term complications can cause serious disability and shortened lifespan. Furthermore, the pancreatic islet β-cell mass has almost completely been destroyed by the time of disease onset, making prediction and prevention a high priority. Also, the increased incidence of T1D, with an average rate of 3% per year [2-5], commands urgency in developing novel prediction and prevention strategies. Biomarkers play essential roles for both identification of high-risk populations and development of prevention strategies.
The etiology of T1D is caused by poorly understood interactions between genetic and environmental factors. The development of the disease involves a cascade of molecular, cellular, and metabolomic impairments which may provide a variety of sources for biomarkers. The long preclinical phase, from genetic susceptibility over active autoimmunity to final overt disease, offers many opportunities for T1D prevention and intervention (Figure 1). Although many T1D prevention trials have been conducted in the last three decades, none of them have proved to be successful. Possible reasons for this failure include heterogeneity of disease pathogenesis, poorly understood etiology, as well as high costs, long periods, and insufficient sample sizes associated with clinical trials.
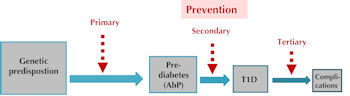 |
 |
Figure 1. Natural history of type 1 diabetes and opportunities for T1D prevention. The natural history of T1D has four main stages: 1. genetic predisposition, 2. prediabetes or autoimmunity, 3. clinical disease onset, and 4. development of diabetic complications. Primary prevention may be applied to the transition from genetic predisposition to autoimmunity. Primary prevention strategies should be highly effective, low-cost and no side effect and can be applied to large numbers of individuals that have increased genetic risk. Secondary prevention can be applied to prediabetic subjects with islet antibodies and the strategies of secondary prevention should be effective and moderate-cost (for example: antigen-based immunization and immunosuppressive drugs). Tertiary prevention is mainly applied to T1D patients with high risk for diabetic complications and the strategies of this step should be effective and reasonable-cost. |
|
Further reasons for the little success in revealing T1D etiology and developing therapies are the lack of suitable biomarkers for the identification and stratification of the high-risk population for specific intervention, and the lack of surrogate biomarkers to evaluate the efficacy of intervention. The combination of genetic susceptibility and islet autoantibody tests has proven to have good predictive value in current trials, although there are major limitations [6]. The specificity of risk identification is generally high for the high-risk categories of individuals, but is low in the general population. More importantly, the appearance of islet autoantibodies, representing active autoimmunity, marks a relatively late stage in disease development. Because T1D prevention may be more effective before an active autoimmune response, biomarkers that can identify the events prior to the appearance of islet autoantibodies would be more valuable. Furthermore, genetic susceptibility and islet autoantibodies cannot be used as surrogate markers for assessing therapeutic outcomes.
During the last three decades, considerable effort has been put into the discovery of novel T1D biomarkers in the genetic, transcriptomic, proteomic, cellular, and metabolomic compartments. The development of high-throughput technologies, especially in genetic, transcriptomic, and proteomic areas, has provided excellent platforms for the discovery of new biomarkers by allowing a systematic coverage of molecular changes during disease progression. In this article, we mainly review progress, opportunities, challenges and potential solutions for these challenges in novel T1D biomarker discovery using these three "omic" methodologies (transcriptomic, proteomic, and metabolomic).
2. Prediction of T1D risk using genetic markers
Genetic factors play an important role in T1D pathogenesis [7]. The strong genetic contribution to T1D is illustrated by the increased risk in siblings (5% by age 20) versus the general population (0.3%) [8] and the high concordance rate in identical twins (up to 65% by age 60) [9]. The search for T1D-associated genes started in the 1970s using primarily two approaches:
1. Linkage studies (using pairs of affected relatives, typically siblings)
2. Association studies (using either case-control or family-based designs)
Although linkage studies have revealed the major contribution of major histocompatibility complex (MHC) to T1D, most non-MHC risk loci (such as insulin (INS), cytotoxic T lymphocyte antigen 4 (CTLA4), protein tyrosine phosphatase, non-receptor type 22 (PTPN22), and interleukin 2 receptor alpha (IL2RA)) were identified using candidate-gene association studies. Recently, association studies have evolved from candidate genes to genome-wide association studies (GWAS), which took advantage of the high-throughput SNP genotyping platforms.
The human leukocyte antigen (HLA) genes were the earliest and crucial findings in the field of T1D genetics. These genes account for approximately 50% of the family clustering [10-13]. With multiple roles in immune reaction, such as T cell selection and antigen presentation, HLA genes can influence disease risk and progression in many ways and can be considered the first checkpoint in the selection and activation of autoimmunity. Subsequent candidate gene studies have identified and confirmed other risk loci. INS [14-17] and PTPN22 [18-22] are two genes with a relative risk of >2.0. INS is a major T1D autoantigen [23, 24], and PTPN22 encodes the lymphoid protein tyrosine phosphatase [25]. CTLA4 [26, 27] and IL2RA [28, 29] are T cell-related genes associated with T1D susceptibility. In addition, the association of T1D with a coding allele of the interferon-induced helicase c domain-containing protein 1 (IFIH1) has been revealed by genome-wide association studies [30, 31]. IFIH1 plays a role in innate immunity through the recognition of the RNA genomes of picornaviruses, providing a potential link between genetic susceptibility and environmental triggers since one of the proposed environmental triggers of T1D, coxsackievirus B4, belongs to the picornavirus family.
Since 2007, highly dense panels of single nucleotide polymorphisms (SNPs) (>300,000 SNPs) distributed across the human genome have been used in GWAS [32-35]. These high-density GWAS confirmed the previously identified loci such as INS, PTPN22, CTLA4, and IL2RA, but more importantly, they provided evidence for a number of novel loci. Surprisingly, the effect sizes estimated for these novel loci were much lower than those for HLA genes, INS and PTPN22, despite the strong statistical power of the GWAS. In view of the large cohort sizes and genome-wide SNP coverage in these studies, it is extremely unlikely that additional loci with large effect can be identified using similar approaches. Furthermore, few of these GWAS loci have yet been mapped to a specific variant or even to a specific gene. Summary results of the GWAS and meta-analysis are available through http://t1dbase.org/.
The advantages of genetic variants as biomarkers are apparent. As germ-line factors, genetic risk variants can serve as a potential predictive tool at a very early stage, even in uterus. Additionally, these genetic factors are relatively easy, inexpensive, and noninvasive to measure. However, there are many challenges in translating these genetic findings into clinical applications. First, genotyping of HLA loci, combined with family history and autoantibody presence, is a current approach for T1D risk prediction with high specificity for the high-risk categories of individuals. However, it has low overall sensitivity in the general population, as most of the T1D cases occur in populations with low or moderate risk. Second, most non-HLA loci have only modest or low individual effects on risk, which hardly increase the predictive value, even if using multiple genetic markers. Third, despite the increasing number of potential target genes, a considerable lack of understanding remains regarding the roles of these genes in the pathogenesis of T1D, and the most efficient ways of application for disease prediction and prevention.
Future genetic studies need to be designed to overcome these challenges. One strategy is to perform follow-up studies for GWAS to better understand the involvement of the candidate loci. The first step to determine the responsible gene(s) and allele(s) is a fine mapping of the regions. Once a reliable risk variant has been identified, the next step is to determine the immediate effects of the gene on gene and protein expression, and the assignable phenotypes in patients with T1D. The second strategy is to perform prospective cohort studies to further validate the GWAS findings and to identify novel genetic factors with larger effect size. All susceptibility genes identified so far are derived from cross-sectional studies that include samples from heterogeneous populations. We anticipate that prospective cohorts are much more suitable for gene mapping studies in complex diseases.
3. Transcriptomic biomarkers for T1D
The dynamic state of the transcriptome during prediabetes, disease progression, and clinical treatment can provide potential biomarkers for disease risk prediction, disease subtype classification, and therapeutic monitoring. Microarray technology and bioinformatics allow the analysis of the whole transcriptome (gene expression profile) in a single experiment. This approach has been successfully applied in many studies, especially on cancer. For example, gene expression profiling has been used to distinguish acute myeloid and acute lymphoblastic leukemia cells [36], to predict outcomes in breast and ovarian cancers [37, 38]. It has also been used to classify subtypes of diffuse large B-cell lymphomas for prognostic implications [39, 40].
A number of studies have attempted to discover changes of gene expression profiles during T1D development [41-48]. Our group identified over 100 genes upregulated in peripheral blood mononuclear cells (PBMCs) of T1D subjects. Most of these genes are also upregulated in prediabetic subjects, suggesting that they may be useful predictive markers [46]. Many of the differentially expressed genes are involved in important immunological functions, including antigen processing and presentation, cytotoxicity and apoptosis (e.g. GZMB, GNLY), and immune regulation (e.g. TGFβ1, SELL). It was found that several proinflammatory mediators and markers (e.g. S100A8/9, NFκB) are upregulated in diabetic and prediabetic subjects. Kaizer and colleagues found overexpression of IL-1-regulated genes as well as chemotaxis and signaling genes in T1D PBMCs [48]. Transcripts corresponding to genes encoding proteins involved in apoptosis and the cell cycle were downregulated in some studies [48], but upregulated in others [46]. Recently, the expression profile for whole blood has suggested an increase in INF-responsive genes at the prediabetic stage [49], a pathway also altered in other autoimmune diseases such as systemic lupus erythematosus (SLE) [50] and Sjogren’s Syndrome [51]. Gene expression patterns were further analyzed in subgroups of patients and controls. Significant differences in gene sets were observed between healthy first-degree relatives of T1D and healthy controls [52], long-term T1D patients and new-onset T1D [45], and juvenile-onset and adult-onset T1D [41]. In addition to the analysis of fresh PBMCs, expression changes inducible in PBMCs cultured with sera from T1D patients have identified soluble factors associated with T1D [53].
In the last decade, much effort has been devoted to discover gene expression patterns in human T1D. However, these studies had the following serious limitations, which affect the validity of their results:
1. Most studies sampled PBMCs rather than pancreatic islets due to the difficulties in obtaining pancreatic samples from human subjects. Transcriptional regulation in the periphery could not be an accurate reflection of autoimmune response in the islets, given the low percentage of islet-reactive lymphocytes in peripheral blood. Also, changes in gene expression that are confined to a particular cell type (e.g. regulatory T cells, dendritic cells and monocyte) may be difficult to detect in PBMCs.
2. The results of most published studies are largely inconclusive and sometimes contradictory. This may be explained by the following reasons: (i) Microarray-based gene expression profiling is a powerful discovery platform, but the results need to be validated by an alternative technique such as real-time polymerase chain reaction (RT-PCR). Unfortunately, few of the previous microarray studies on T1D have been further confirmed by a validation study. (ii) Most previous studies had very small sample sizes (less than 100 subjects in each group) which are not adequate for the human population given the large expression variations among individual subjects.
3. Most gene expression profile data were derived from cross-sectional studies, which is a good approach for biomarker discovery, but not good enough for biomarker validation. There were a few studies using longitudinal samples to characterize the gene expression signatures during T1D progression; however, the small sample size limited their study power [44]. Therefore, prospective studies with large cohorts need to be designed for future studies.
Transcriptomic biomarkers for T1D are still not ready for clinical application even though this approach holds great promise. One urgent task is the validation of the previous findings from microarray data using an alternative method such as RT-PCR. To obtain more reliable and reproducible results, the experimental design needs to be improved in the following ways:
1. Use of large sample size, at least a thousand in each group to get consistent results based on our validation data (Table 1), which showed inconsistent data for some genes between two independent medium-sized sample sets.
2. When expression data are generated on multiple 384-well plates for thousands of samples, plate-to-plate variation should be recognized and normalized to ensure consistency across plates.
3. Given the huge intra-group individual variation, only looking at fold changes of gene expression among studied groups, as done in most previous studies, is not enough to identify T1D associated genes. Other statistical approaches, such as conditional logistic regression, should also be used to estimate the relative risk.
4. Selection of appropriate reference genes for normalization of quantitative Real-Time PCR has a major impact on data quality [54-57]. Most of the previous studies have used only a single reference gene for normalization. To avoid biased results, gene transcription studies using RT-PCR should begin with the selection of an appropriate set of reference genes.
Rapid progress is being made in the development of novel technologies for transcriptomics. Notably, the introduction of high-throughput next-generation DNA sequencing (NGS) technologies revolutionized transcriptomics by performing RNA analysis through cDNA sequencing on a massive scale [58]. This development eliminated several challenges associated with microarray technologies, and has provided a better knowledge of both quantitative and qualitative aspects of transcriptomics. This novel technology allows a more comprehensive understanding of transcription initiation sites, the cataloguing of sense and antisense transcripts, previously unknown coding and non-coding RNA species, particularly small RNAs (e.g. micro RNA), improved detection of alternative splicing events, and improved detection of gene fusion transcripts [59]. We believe that this technology will become increasingly important in T1D research, and provide unparalleled opportunities for biomarker discovery.
Table
1.
Summary of gene expression validation data |
|
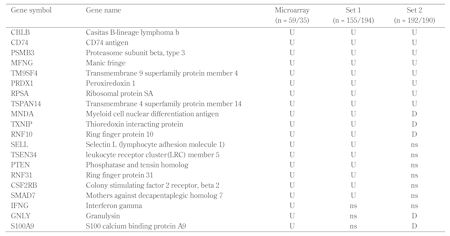 |
|
Legend:
U – upregulated in T1D; D – downregulated in T1D; ns – not significant. |
|
4. Proteomic biomarkers for T1D
Protein levels execute the aberrant genetic and genomic changes, and therefore are more directly correlated with cellular function and health status. Thus, they have a greater potential to be used as biomarkers for disease prediction or therapeutic monitoring. Today, the most useful proteomic biomarkers for T1D are "islet autoantibodies" present in serum, which are strong predictors of the later development of T1D. Since the 1970s, a series of islet autoantibodies (over 20) involved in T1D has been discovered [60-79]. Autoantibody assays have constantly been improved. The four major autoantibodies of clinical and research interest are islet cell cytoplasmic autoantibodies (ICA), glutamic acid decarboxylase autoantibodies (GADA), insulinoma 2-associated autoantibodies (IA-2A), and insulin autoantibodies (IAA). ZnT8A, a newly recognized ZnT8 islet autoantibody, may further improve the value of islet autoantibody testing. Other identified autoantibodies are either difficult to measure and/or are not sufficiently sensitive or specific to enable their use as T1D markers [80]. Although the major islet autoantibodies are not considered to be involved in the pathogenesis of T1D, they are the hallmark of autoimmune response to autoantigens, and they are critical for the design of clinical trials for T1D prevention [6].
Unquestionably, the risk of developing T1D will rise with the increase in number of islet autoantibodies present. The risk for T1D in individuals without any autoantibody is only 0.5%, and rise to approximately 3% in individuals with one autoantibody. In subjects with two autoantibodies the risk keeps rising to 16%, and jumps to 40% and 50% in subjects with three and four autoantibodies, respectively [81]. IAA is less predictive of T1D than other autoantibodies. Higher ICA titers and higher concentrations of GADA were more powerful predictors of T1D than lower titers and lower concentrations. Despite the usage of the autoantibodies in T1D prediction, they have several serious limitations:
1. The appearance of islet autoantibodies marks a relatively late stage of the autoimmune process, and therefore is not suitable for early disease intervention. A recent study suggested that some extrapancreatic autoantibodies were present prior to detection of islet autoantibodies; however, their potential for biomarkers need to be further assessed [82].
2. Only a subset of the autoantibody-positive subjects will progress to clinical diabetes. Therefore, it would be desirable to have biomarkers that allow the distinction of the progressors versus non-progressors.
3. Autoantibodies are not useful as biomarkers for therapeutic outcomes.
Other than autoantibodies, several immune molecules, like cytokines and chemokines, have been widely studied for their potential roles in T1D development and for the possibility as T1D biomarkers. The current view is that T1D arises from T cell-mediated islet cell destruction initiated by an imbalance in Th1 and Th2 cells [83]. T1D is believed to be mediated by Th1 cytokines. Several studies have found higher serum levels of Th1 cytokines in diabetic patients and their first-degree relatives compared to healthy controls [84-88]. A similar pattern of Th1/2 cytokine profile between newly diagnosed T1D patients and their healthy siblings was further confirmed in a recent study with a relatively large sample size (500 subjects in each group) [89]. The Th1 cytokines IL-1β, TNF-α, and IFN-γ have been shown to be cytotoxic to β-cells by inducing nitric oxide (NO) production [90]. Some studies suggested a significantly higher IL-1 production but a lower IL-1Ra (IL1 receptor antagonist) in newly diagnosed T1D patients compared with chronic T1D patients [91, 92].
MCP-1 and IP-10 are the two best studied chemokines in T1D being chemo-attractors for monocytes and activated Th1 and NK cells specifically. Studies with animal models have demonstrated that high levels of MCP-1 and IP-10 are released by islets cells during autoimmune attack [93-96]. Serum levels of these two cytokines have been measured in T1D patients and healthy controls in several studies. However, all these studies had extremely small sample sizes, and the conclusions from these studies were inconclusive and inconsistent. A study by our group has measured serum MCP-1 in a large cohort (with 2724 T1D patients and 2654 controls) [97]. Interestingly, serum MCP-1 levels were significantly higher in patients with multiple complications than in patients without any complications. They were also higher in controls than T1D patients, which suggested MCP-1 may have a dual role in T1D and its complications. Currently, assay sensitivity is a major limitation for the accurate measurement of many cytokines and chemokines with low concentration, such as GM-CSF, INF-γ, and interleukins [98].
Quantitative analysis of global protein levels is important for the systematic understanding of the molecular changes associated with disease progression, and may provide insight into disease pathogenesis and management. Recent development of mass spectrometry (MS)-based technology has provided useful platforms for the study of quantitative changes in protein components [99, 100]. Several methods are widely used in proteomic analysis, including two-dimensional (2D) gel electrophoresis followed by MS analysis, MS signal intensity-based quantification, stable isotope labeling-based quantification, and intact protein-based quantification [101-103]. Because of the difficulties in obtaining pathological tissues from T1D patients due to ethical and practical concerns, serum is an excellent alternative resource for biomarker discovery. It is rich in biological information and easy available. However, comprehensive analysis of the serum proteome is a challenging task due to its extraordinary complexity and high dynamic range in concentration. The complexity of serum proteome results from its charge, molecular mass, and hydrophobicity, as well as its expression level and post-translational modifications. Only abundant proteins are analyzable by currently available methods due to their high dynamic range in protein concentration. Unfortunately, low abundance proteins that are promising biomarkers are difficult to detect and quantify. This difficulty is particularly true for serum because more than 99% of the proteins consist of serum albumin and globulins.
Therefore, sample pretreatment is required to enrich the low- to medium-abundance proteins, called protein normalization. Our group compared different approaches for low-abundance enrichment, and showed that random hexapeptide library beads have distinct advantages over the traditional immune-depletion methods due to their higher efficiency, higher binding capacity, and lower costs [104]. We also evaluated in-depth mining of serum/plasma proteome using different separation techniques. Our data suggested that shotgun proteomics—multidimensional separation of digested peptides followed by mass spectrometry analysis—is highly efficient, and therefore should be the desirable method for protein biomarker discovery [105, 106]. Using these preferred strategies, our group, as one of the pioneers, systematically discovered and validated serum proteomic changes in T1D patients [106]. We found that two well-known inflammation mediators, serum amyloid protein A (SAA) and C-reactive protein (CRP), as well as adiponectin and insulin-like growth factor binding protein 2 have significantly higher serum levels in T1D patients. Whereas, there are lower serum levels for two other proteins: transforming growth factor beta induced (TGFβI) and myeloperoxidase. In particular, the subjects in the top quartile for expression of these markers had the highest risk of T1D (relative risk is up to 10).
In addition to serum, urine represents another excellent specimen for proteome analysis due to its easy availability and higher stability than blood [107]. Maahs and colleagues discovered and further validated urinary proteomic biomarkers for diabetes in general, and for specific type of diabetes [108]. The difference of urinary collagen fragments between T1D and T2D suggested that different mechanisms of extracellular matrix remodeling exist in these two types of diabetes.
In summary, despite considerable progress in proteomic biomarker discovery for T1D, no new "star protein biomarkers" of equal importance to islet autoantibodies have been identified to date, due to both technical and biological issues, as aforementioned. Proteomic technology needs to be improved in several areas, including quantification and identification to low-abundance proteins, assessment of protein distribution among cells and subcellular compartments, and assessment of post-translational modification. In addition to the need for technical improvement, several issues need to be considered for better study design. One is the appropriate selection of biological specimens for each purpose. For example, serum probably is the most suitable specimen for T1D biomarker discovery due to its high richness of proteins, while urinary samples may be more suitable for T1D complication studies, especially for diabetic nephropathy. Furthermore, it should be recognized that no single analytical technique is suited to address the proteomic complexities. For example, the MS platform is suitable for the discovery phase, but not for validation. Finally, reasonable sample size, in both cross-sectional and longitudinal studies, is a key factor for the identification and validation of reliable T1D biomarkers.
5. Conclusions
The long asymptomatic period of T1D provides many opportunities for disease prevention and intervention. Genetic susceptibility and islet autoantibodies are still the most useful biomarkers for T1D risk prediction. However, these currently available markers do not fully meet the need for T1D prediction and prevention due to their low predictive value and relative late appearance.
Increasing efforts have been devoted to novel T1D biomarker discovery in the last three decades. The most noteworthy advances are developments of high throughput "omic" technologies, which offer great opportunities for biomarker discovery. Unfortunately, so far "star biomarkers" with the potential to better predict T1D risk or evaluate therapeutic outcomes have not been discovered because of both biological and technical challenges. To overcome these issues, improvements in technologies and study design need to be made in future studies.
Disclosure: The authors report no conflict of interests.
Acknowledgments:
This work is partly supported by grants from the National Institutes of Health (4R33HD050196, 4R33DK069878, and 2RO1HD37800) and JDRF (1-2004-661) given to JXS.
References
- Atkinson MA, Maclaren NK. The pathogenesis of insulin-dependent diabetes mellitus. N Engl J Med 1994. 331(21):1428-1436. [DOD] [CrossRef]
- Borchers AT, Uibo R, Gershwin ME. The geoepidemiology of type 1 diabetes. Autoimmun Rev 2010. 9(5):A355-A365. [DOD] [CrossRef]
- Bruno G, Novelli G, Panero F, Perotto M, Monasterolo F, Bona G, Perino A, Rabbone I, Cavallo-Perin P, Cerutti F. The incidence of type 1 diabetes is increasing in both children and young adults in Northern Italy: 1984-2004 temporal trends. Diabetologia 2009. 52(12):2531-2535. [DOD] [CrossRef]
- Chan JC, Malik V, Jia W, Kadowaki T, Yajnik CS, Yoon KH, Hu FB. Diabetes in Asia: epidemiology, risk factors, and pathophysiology. JAMA 2009. 301(20):2129-2140. [DOD] [CrossRef]
- Levitt NS. Diabetes in Africa: epidemiology, management and healthcare challenges. Heart 2008. 94(11):1376-1382. [DOD] [CrossRef]
- Ziegler AG, Nepom GT. Prediction and pathogenesis in type 1 diabetes. Immunity 2010. 32(4):468-478. [DOD] [CrossRef]
- Concannon P, Rich SS, Nepom GT. Genetics of type 1A diabetes. N Engl J Med 2009. 360(16):1646-1654. [DOD] [CrossRef]
- Bonifacio E, Ziegler AG. Advances in the prediction and natural history of type 1 diabetes. Endocrinol Metab Clin North Am 2010. 39(3):513-525. [DOD] [CrossRef]
- Redondo MJ, Jeffrey J, Fain PR, Eisenbarth GS, Orban T. Concordance for islet autoimmunity among monozygotic twins. N Engl J Med 2008. 359(26):2849-2850. [DOD] [CrossRef]
- Cudworth AG, Woodrow JC. Evidence for HL-A-linked genes in "juvenile" diabetes mellitus. Br Med J 1975. 3(5976):133-135. [DOD] [CrossRef]
- Nerup J, Platz P, Andersen OO, Christy M, Lyngsoe J, Poulsen JE, Ryder LP, Nielsen LS, Thomsen M, Svejgaard A. HL-A antigens and diabetes mellitus. Lancet 1974. 2(7885):864-866. [DOD] [CrossRef]
- Singal DP, Blajchman MA. Histocompatibility (HL-A) antigens, lymphocytotoxic antibodies and tissue antibodies in patients with diabetes mellitus. Diabetes 1973. 22(6):429-432. [DOD]
- Risch N. Assessing the role of HLA-linked and unlinked determinants of disease. Am J Hum Genet 1987. 40(1):1-14. [DOD]
- Bell GI, Horita S, Karam JH. A polymorphic locus near the human insulin gene is associated with insulin-dependent diabetes mellitus. Diabetes 1984. 33(2):176-183. [DOD] [CrossRef]
- Lucassen AM, Julier C, Beressi JP, Boitard C, Froguel P, Lathrop M, Bell JI. Susceptibility to insulin dependent diabetes mellitus maps to a 4.1 kb segment of DNA spanning the insulin gene and associated VNTR. Nat Genet 1993. 4(3):305-310. [DOD] [CrossRef]
- Bennett ST, Lucassen AM, Gough SC, Powell EE, Undlien DE, Pritchard LE, Merriman ME, Kawaguchi Y, Dronsfield MJ, Pociot F, et al. Susceptibility to human type 1 diabetes at IDDM2 is determined by tandem repeat variation at the insulin gene minisatellite locus. Nat Genet 1995. 9(3):284-292. [DOD] [CrossRef]
- Barratt BJ, Payne F, Lowe CE, Hermann R, Healy BC, Harold D, Concannon P, Gharani N, McCarthy MI, Olavesen MG, et al. Remapping the insulin gene/IDDM2 locus in type 1 diabetes. Diabetes 2004. 53(7):1884-1889. [DOD] [CrossRef]
- Bottini N, Musumeci L, Alonso A, Rahmouni S, Nika K, Rostamkhani M, MacMurray J, Meloni GF, Lucarelli P, Pellecchia M, Eisenbarth GS, Comings D, Mustelin T. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat Genet 2004. 36(4):337-338. [DOD] [CrossRef]
- Smyth D, Cooper JD, Collins JE, Heward JM, Franklyn JA, Howson JM, Vella A, Nutland S, Rance HE, Maier L, et al. Replication of an association between the lymphoid tyrosine phosphatase locus (LYP/PTPN22) with type 1 diabetes, and evidence for its role as a general autoimmunity locus. Diabetes 2004. 53(11):3020-3023. [DOD] [CrossRef]
- Qu H, Tessier MC, Hudson TJ, Polychronakos C. Confirmation of the association of the R620W polymorphism in the protein tyrosine phosphatase PTPN22 with type 1 diabetes in a family based study. J Med Genet 2005. 42(3):266-270. [DOD] [CrossRef]
- Onengut-Gumuscu S, Ewens KG, Spielman RS, Concannon P. A functional polymorphism (1858C/T) in the PTPN22 gene is linked and associated with type I diabetes in multiplex families. Genes Immun 2004. 5(8):678-680. [DOD] [CrossRef]
- Ladner MB, Bottini N, Valdes AM, Noble JA. Association of the single nucleotide polymorphism C1858T of the PTPN22 gene with type 1 diabetes. Hum Immunol 2005. 66(1):60-64. [DOD] [CrossRef]
- Kent SC, Chen Y, Bregoli L, Clemmings SM, Kenyon NS, Ricordi C, Hering BJ, Hafler DA. Expanded T cells from pancreatic lymph nodes of type 1 diabetic subjects recognize an insulin epitope. Nature 2005. 435(7039):224-228. [DOD] [CrossRef]
- Nakayama M, Abiru N, Moriyama H, Babaya N, Liu E, Miao D, Yu L, Wegmann DR, Hutton JC, Elliott JF, Eisenbarth GS. Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice. Nature 2005. 435(7039):220-223. [DOD] [CrossRef]
- Kyogoku C, Langefeld CD, Ortmann WA, Lee A, Selby S, Carlton VE, Chang M, Ramos P, Baechler EC, Batliwalla FM, et al. Genetic association of the R620W polymorphism of protein tyrosine phosphatase PTPN22 with human SLE. Am J Hum Genet 2004. 75(3):504-507. [DOD] [CrossRef]
- Nistico L, Buzzetti R, Pritchard LE, Van der Auwera B, Giovannini C, Bosi E, Larrad MT, Rios MS, Chow CC, Cockram CS, et al. The CTLA-4 gene region of chromosome 2q33 is linked to, and associated with, type 1 diabetes. Belgian Diabetes Registry. Hum Mol Genet 1996. 5(7):1075-1080. [DOD] [CrossRef]
- Ueda H, Howson JM, Esposito L, Heward J, Snook H, Chamberlain G, Rainbow DB, Hunter KM, Smith AN, Di Genova G, et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature 2003. 423(6939):506-511. [DOD] [CrossRef]
- Vella A, Cooper JD, Lowe CE, Walker N, Nutland S, Widmer B, Jones R, Ring SM, McArdle W, Pembrey ME, et al. Localization of a type 1 diabetes locus in the IL2RA/CD25 region by use of tag single-nucleotide polymorphisms. Am J Hum Genet 2005. 76(5):773-779. [DOD] [CrossRef]
- Lowe CE, Cooper JD, Brusko T, Walker NM, Smyth DJ, Bailey R, Bourget K, Plagnol V, Field S, Atkinson M, Clayton DG, Wicker LS, Todd JA. Large-scale genetic fine mapping and genotype-phenotype associations implicate polymorphism in the IL2RA region in type 1 diabetes. Nat Genet 2007. 39(9):1074-1082. [DOD] [CrossRef]
- Smyth DJ, Cooper JD, Bailey R, Field S, Burren O, Smink LJ, Guja C, Ionescu-Tirgoviste C, Widmer B, Dunger DB, Savage DA, Walker NM, Clayton DG, Todd JA. A genome-wide association study of nonsynonymous SNPs identifies a type 1 diabetes locus in the interferon-induced helicase (IFIH1) region. Nat Genet 2006. 38(6):617-619. [DOD] [CrossRef]
- Liu S, Wang H, Jin Y, Podolsky R, Reddy MV, Pedersen J, Bode B, Reed J, Steed D, Anderson S, et al. IFIH1 polymorphisms are significantly associated with type 1 diabetes and IFIH1 gene expression in peripheral blood mononuclear cells. Hum Mol Genet 2009. 18(2):358-365. [DOD] [CrossRef]
- Polychronakos C, Li Q. Understanding type 1 diabetes through genetics: advances and prospects. Nat Rev Genet 2011. 12(11):781-792. [DOD] [CrossRef]
- Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, Plagnol V, Bailey R, Nejentsev S, Field SF, Payne F, et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet 2007. 39(7):857-864. [DOD] [CrossRef]
- Cooper JD, Smyth DJ, Smiles AM, Plagnol V, Walker NM, Allen JE, Downes K, Barrett JC, Healy BC, Mychaleckyj JC, Warram JH, Todd JA. Meta-analysis of genome-wide association study data identifies additional type 1 diabetes risk loci. Nat Genet 2008. 40(12):1399-1401. [DOD] [CrossRef]
- Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, Julier C, Morahan G, Nerup J, Nierras C, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet 2009. 41(6):703-707. [DOD] [CrossRef]
- Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caligiuri MA, Bloomfield CD, Lander ES. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science 1999. 286(5439):531-537. [DOD] [CrossRef]
- Berchuck A, Iversen ES, Lancaster JM, Pittman J, Luo J, Lee P, Murphy S, Dressman HK, Febbo PG, West M, Nevins JR, Marks JR. Patterns of gene expression that characterize long-term survival in advanced stage serous ovarian cancers. Clin Cancer Res 2005. 11(10):3686-3696. [DOD] [CrossRef]
- Huang E, Cheng SH, Dressman H, Pittman J, Tsou MH, Horng CF, Bild A, Iversen ES, Liao M, Chen CM, West M, Nevins JR, Huang AT. Gene expression predictors of breast cancer outcomes. Lancet 2003. 361(9369):1590-1596. [DOD] [CrossRef]
- Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000. 403(6769):503-511. [DOD] [CrossRef]
- Tirado CA, Chen W, Garcia R, Kohlman KA, Rao N. Genomic profiling using array comparative genomic hybridization define distinct subtypes of diffuse large b-cell lymphoma: a review of the literature. J Hematol Oncol 2012. 5:54. [DOD] [CrossRef]
- Padmos RC, Schloot NC, Beyan H, Ruwhof C, Staal FJ, de Ridder D, Aanstoot HJ, Lam-Tse WK, de Wit H, de Herder C, et al. Distinct monocyte gene-expression profiles in autoimmune diabetes. Diabetes 2008. 57(10):2768-2773. [DOD] [CrossRef]
- Kaizer EC, Glaser CL, Chaussabel D, Banchereau J, Pascual V, White PC. Gene expression in peripheral blood mononuclear cells from children with diabetes. J Clin Endocrinol Metab 2007. 92(9):3705-3711. [DOD] [CrossRef]
- Planas R, Carrillo J, Sanchez A, de Villa MC, Nunez F, Verdaguer J, James RF, Pujol-Borrell R, Vives-Pi M. Gene expression profiles for the human pancreas and purified islets in type 1 diabetes: new findings at clinical onset and in long-standing diabetes. Clin Exp Immunol 2010. 159(1):23-44. [DOD] [CrossRef]
- Elo LL, Mykkanen J, Nikula T, Jarvenpaa H, Simell S, Aittokallio T, Hyöty H, Ilonen J, Veijola R, Simell T, Knip M, Simell O, Lahesmaa R. Early suppression of immune response pathways characterizes children with prediabetes in genome-wide gene expression profiling. J Autoimmun 2010. 35(1):70-76. [DOD] [CrossRef]
- Han D, Cai X, Wen J, Matheson D, Skyler JS, Kenyon NS, Chen Z. Innate and adaptive immune gene expression profiles as biomarkers in human type 1 diabetes. Clin Exp Immunol 2012. 170(2):131-138. [DOD] [CrossRef]
- Collins CD, Purohit S, Podolsky RH, Zhao HS, Schatz D, Eckenrode SE, Yang P, Hopkins D, Muir A, Hoffman M, McIndoe RA, Rewers M, She JX. The application of genomic and proteomic technologies in predictive, preventive and personalized medicine. Vascul Pharmacol 2006. 45(5):258-267. [DOD] [CrossRef]
- Planas R, Pujol-Borrell R, Vives-Pi M. Global gene expression changes in type 1 diabetes: insights into autoimmune response in the target organ and in the periphery. Immunol Lett 2010. 133(2):55-61. [DOD] [CrossRef]
- Kaizer EC, Glaser CL, Chaussabel D, Banchereau J, Pascual V, White PC. Gene expression in peripheral blood mononuclear cells from children with diabetes. J Clin Endocrinol Metab 2007. 92(9):3705-3711. [DOD] [CrossRef]
- Reynier F, Pachot A, Paye M, Xu Q, Turrel-Davin F, Petit F, Hot A, Auffray C, Bendelac N, Nicolino M, Mougin B, Thivolet C. Specific gene expression signature associated with development of autoimmune type-I diabetes using whole-blood microarray analysis. Genes Immun 2010. 11(3):269-278. [DOD] [CrossRef]
- Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, Shark KB, Grande WJ, Hughes KM, Kapur V, Gregersen PK, Behrens TW. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A 2003. 100(5):2610-2615. [DOD] [CrossRef]
- Emamian ES, Leon JM, Lessard CJ, Grandits M, Baechler EC, Gaffney PM, Segal B, Rhodus NL, Moser KL. Peripheral blood gene expression profiling in Sjogren's syndrome. Genes Immun 2009. 10(4):285-296. [DOD] [CrossRef]
- Stechova K, Kolar M, Blatny R, Halbhuber Z, Vcelakova J, Hubackova M, Petruzelkova L, Sumnik Z, Obermannova B, Pithova P, et al. Healthy first degree relatives of patients with type 1 diabetes exhibit significant differences in basal gene expression pattern of immunocompetent cells compared to controls: expression pattern as predeterminant of autoimmune diabetes. Scand J Immunol 2011. In press. [DOD]
- Wang X, Jia S, Geoffrey R, Alemzadeh R, Ghosh S, Hessner MJ. Identification of a molecular signature in human type 1 diabetes mellitus using serum and functional genomics. J Immunol 2008. 180(3):1929-1937. [DOD]
- Chervoneva I, Li Y, Schulz S, Croker S, Wilson C, Waldman SA, Hyslop T. Selection of optimal reference genes for normalization in quantitative RT-PCR. BMC Bioinformatics 2010. 11:253. [DOD] [CrossRef]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 2002. 3(7):RESEARCH0034. [DOD] [CrossRef]
- Stamova BS, Apperson M, Walker WL, Tian Y, Xu H, Adamczy P, Zhan X, Liu DZ, Ander BP, Liao IH, et al. Identification and validation of suitable endogenous reference genes for gene expression studies in human peripheral blood. BMC Med Genomics 2009. 2:49. [DOD] [CrossRef]
- Dheda K, Huggett JF, Chang JS, Kim LU, Bustin SA, Johnson MA, Rook GA, Zumla A. The implications of using an inappropriate reference gene for real-time reverse transcription PCR data normalization. Anal Biochem 2005. 344(1):141-143. [DOD] [CrossRef]
- Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet 2010. 11(1):31-46. [DOD] [CrossRef]
- Ozsolak F, Milos PM. RNA sequencing: advances, challenges and opportunities. Nat Rev Genet 2011. 12(2):87-98. [DOD] [CrossRef]
- Palmer JP, Asplin CM, Clemons P, Lyen K, Tatpati O, Raghu PK, Paquette TL. Insulin antibodies in insulin-dependent diabetics before insulin treatment. Science 1983. 222(4630):1337-1339. [DOD] [CrossRef]
- Kuglin B, Halder B, Bertrams J, Gruneklee D, Gries FA, Kolb H. Proinsulin autoantibodies: association with type I diabetes but not with islet cell antibodies, insulin autoantibodies or HLA-DR type. J Autoimmun 1990. 3(5):573-577. [DOD] [CrossRef]
- Wenzlau JM, Moua O, Sarkar SA, Yu L, Rewers M, Eisenbarth GS, Davidson HW, Hutton JC. SlC30A8 is a major target of humoral autoimmunity in type 1 diabetes and a predictive marker in prediabetes. Ann N Y Acad Sci 2008. 1150:256-259. [DOD] [CrossRef]
- Verge CF, Gianani R, Kawasaki E, Yu L, Pietropaolo M, Jackson RA, Chase HP, Eisenbarth GS. Prediction of type I diabetes in first-degree relatives using a combination of insulin, GAD, and ICA512bdc/IA-2 autoantibodies. Diabetes 1996. 45(7):926-933. [DOD] [CrossRef]
- Kawasaki E, Hutton JC, Eisenbarth GS. Molecular cloning and characterization of the human transmembrane protein tyrosine phosphatase homologue, phogrin, an autoantigen of type 1 diabetes. Biochem Biophys Res Commun 1996. 227(2):440-447. [DOD] [CrossRef]
- Taniguchi T, Okazaki K, Okamoto M, Seko S, Uchida K, Seino Y. Presence of autoantibodies to carbonic anhidrase II and lactoferrin in type 1 diabetes: proposal of the concept of autoimmune exocrinopathy and endocrinopathy of the pancreas. Diabetes Care 2001. 24(9):1695-1696. [DOD] [CrossRef]
- Kim YJ, Zhou Z, Hurtado J, Wood DL, Choi AS, Pescovitz MD, Warfel KA, Vandagriff J, Davis JK, Kwon BS. IDDM patients' sera recognize a novel 30-kD pancreatic autoantigen related to chymotrypsinogen. Immunol Invest 1993. 22(3):219-227. [DOD] [CrossRef]
- Chang YH, Hwang J, Shang HF, Tsai ST. Characterization of human DNA topoisomerase II as an autoantigen recognized by patients with IDDM. Diabetes 1996. 45(4):408-414. [DOD] [CrossRef]
- Baekkeskov S, Aanstoot HJ, Christgau S, Reetz A, Solimena M, Cascalho M, Folli F, Richter-Olesen H, De Camilli P. Identification of the 64K autoantigen in insulin-dependent diabetes as the GABA-synthesizing enzyme glutamic acid decarboxylase. Nature 1990. 347(6289):151-156. [DOD] [CrossRef]
- Rorsman F, Husebye ES, Winqvist O, Bjork E, Karlsson FA, Kampe O. Aromatic-L-amino-acid decarboxylase, a pyridoxal phosphate-dependent enzyme, is a beta-cell autoantigen. Proc Natl Acad Sci U S A 1995. 92(19):8626-8629. [DOD]
- Park SG, Park HS, Jeong IK, Cho YM, Lee HK, Kang YS, Kim S, Park KS. Autoantibodies against aminoacyl-tRNA synthetase: novel diagnostic marker for type 1 diabetes mellitus. Biomarkers 2010. 15(4):358-366. [DOD] [CrossRef]
- Aanstoot HJ, Kang SM, Kim J, Lindsay LA, Roll U, Knip M, Atkinson M, Mose-Larsen P, Fey S, Ludvigsson J, et al. Identification and characterization of glima 38, a glycosylated islet cell membrane antigen, which together with GAD65 and IA2 marks the early phases of autoimmune response in type 1 diabetes. J Clin Invest 1996. 97(12):2772-2783. [DOD] [CrossRef]
- Cabrera-Rode E, Diaz-Horta O, Fernandez LE, Carr A, Marquina G, Valiente O, Gonzalez-Suarez RM, Uriarte A. Glycolipids as the major autoantigens of cytoplasmatic islet cell antibodies. Autoimmunity 1995. 20(3):145-151. [DOD] [CrossRef]
- Jones DB, Hunter NR, Duff GW. Heat-shock protein 65 as a beta cell antigen of insulin-dependent diabetes. Lancet 1990. 336(8715):583-585. [DOD] [CrossRef]
- Bottazzo GF, Florin-Christensen A, Doniach D. Islet-cell antibodies in diabetes mellitus with autoimmune polyendocrine deficiencies. Lancet 1974. 2(7892):1279-1283. [DOD] [CrossRef]
- Karounos DG, Thomas JW. Recognition of common islet antigen by autoantibodies from NOD mice and humans with IDDM. Diabetes 1990. 39(9):1085-1090. [DOD] [CrossRef]
- Johnson JH, Crider BP, McCorkle K, Alford M, Unger RH. Inhibition of glucose transport into rat islet cells by immunoglobulins from patients with new-onset insulin-dependent diabetes mellitus. N Engl J Med 1990. 322(10):653-659. [DOD] [CrossRef]
- Dotta F, Falorni A, Tiberti C, Dionisi S, Anastasi E, Torresi P, Lernmark A, Di Mario U. Autoantibodies to the GM2-1 islet ganglioside and to GAD-65 at type 1 diabetes onset. J Autoimmun 1997. 10(6):585-588. [DOD] [CrossRef]
- Maclaren NK, Huang SW, Fogh J. Antibody to cultured human insulinoma cells in insulin-dependent diabetes. Lancet 1975. 1(7914):997-1000. [DOD] [CrossRef]
- Pak CY, Cha CY, Rajotte RV, McArthur RG, Yoon JW. Human pancreatic islet cell specific 38 kilodalton autoantigen identified by cytomegalovirus-induced monoclonal islet cell autoantibody. Diabetologia 1990. 33(9):569-572. [DOD] [CrossRef]
- Winter WE, Schatz DA. Autoimmune markers in diabetes. Clin Chem 2011. 57(2):168-175. [DOD] [CrossRef]
- Orban T, Sosenko JM, Cuthbertson D, Krischer JP, Skyler JS, Jackson R, Yu L, Palmer JP, Schatz D, Eisenbarth G. Pancreatic islet autoantibodies as predictors of type 1 diabetes in the Diabetes Prevention Trial-Type 1. Diabetes Care 2009. 32(12):2269-2274. [DOD] [CrossRef]
- Burbelo PD, Lebovitz EE, Bren KE, Bayat A, Paviol S, Wenzlau JM, Barriga KJ, Rewers M, Harlan DM, Iadarola MJ. Extrapancreatic autoantibody profiles in type I diabetes. PLoS One 2012. 7(9):e45216. [DOD]
- Sia C. Imbalance in Th cell polarization and its relevance in type 1 diabetes mellitus. Rev Diabet Stud 2005. 2(4):182-186. [DOD] [CrossRef]
- Nerup J, Mandrup-Poulsen T, Helqvist S, Andersen HU, Pociot F, Reimers JI, Cuartero BG, Karlsen AE, Bjerre U, Lorenzen T. On the pathogenesis of IDDM. Diabetologia 1994. 37(Suppl 2):S82-S89. [DOD] [CrossRef]
- Hussain MJ, Maher J, Warnock T, Vats A, Peakman M, Vergani D. Cytokine overproduction in healthy first degree relatives of patients with IDDM. Diabetologia 1998. 41(3):343-349. [DOD] [CrossRef]
- Rabinovitch A, Suarez-Pinzon WL. Role of cytokines in the pathogenesis of autoimmune diabetes mellitus. Rev Endocr Metab Disord 2003. 4(3):291-299. [DOD] [CrossRef]
- Karlsson MG, Lawesson SS, Ludvigsson J. Th1-like dominance in high-risk first-degree relatives of type I diabetic patients. Diabetologia 2000. 43(6):742-749. [DOD] [CrossRef]
- Kallmann BA, Lampeter EF, Hanifi-Moghaddam P, Hawa M, Leslie RD, Kolb H. Cytokine secretion patterns in twins discordant for Type I diabetes. Diabetologia 1999. 42(9):1080-1085. [DOD] [CrossRef]
- Svensson J, Eising S, Hougaard DM, Mortensen HB, Skogstrand K, Simonsen LB, Carstensen B, Nilsson A, Lernmark A, Pociot F, Johannesen J. Few differences in cytokines between patients newly diagnosed with type 1 diabetes and their healthy siblings. Hum Immunol 2012. 73(11):1116-1126. [DOD] [CrossRef]
- Bergholdt R, Heding P, Nielsen K, Nolsoe R, Sparre T, Storling J, Nerup J, Pociot F, Mandrup-Poulsen T. Type 1 database mellitus: an inflammatory disease of the islet. Adv Exp Med Biol 2004. 552:129-153. [DOD]
- Aribi M, Moulessehoul S, Kendouci-Tani M, Benabadji AB, Hichami A, Khan NA. Relationship between interleukin-1beta and lipids in type 1 diabetic patients. Med Sci Monit 2007. 13(8):CR372-CR378. [DOD]
- Netea MG, Hancu N, Blok WL, Grigorescu-Sido P, Popa L, Popa V, van der Meer JW. Interleukin 1 beta, tumour necrosis factor-alpha and interleukin 1 receptor antagonist in newly diagnosed insulin-dependent diabetes mellitus: comparison to long-standing diabetes and healthy individuals. Cytokine 1997. 9(4):284-287. [DOD] [CrossRef]
- Reddy S, Bai Y, Robinson E, Ross J. Immunolocalization of monocyte chemoattractant protein-1 in islets of NOD mice during cyclophosphamide administration. Ann N Y Acad Sci 2006. 1079:103-108. [DOD] [CrossRef]
- Lee I, Wang L, Wells AD, Ye Q, Han R, Dorf ME, Kuziel WA, Rollins BJ, Chen L, Hancock WW. Blocking the monocyte chemoattractant protein-1/CCR2 chemokine pathway induces permanent survival of islet allografts through a programmed death-1 ligand-1-dependent mechanism. J Immunol 2003. 171(12):6929-6935. [DOD]
- Chipitsyna G, Gong Q, Gray CF, Haroon Y, Kamer E, Arafat HA. Induction of monocyte chemoattractant protein-1 expression by angiotensin II in the pancreatic islets and beta-cells. Endocrinology 2007. 148(5):2198-2208. [DOD] [CrossRef]
- Christen U, von Herrath MG. IP-10 and type 1 diabetes: a question of time and location. Autoimmunity 2004. 37(4):273-282. [DOD] [CrossRef]
- Guan R, Purohit S, Wang H, Bode B, Reed JC, Steed RD, Anderson SW, Steed L, Hopkins D, Xia C, She JX. Chemokine (C-C motif) ligand 2 (CCL2) in sera of patients with type 1 diabetes and diabetic complications. Plos One 2011. 6(4):e17822. [DOD] [CrossRef]
- Purohit S, She JX. Biomarkers for type 1 diabetes. Int J Clin Exp Med 2008. 1(2):98-116. [DOD]
- Elias JE, Haas W, Faherty BK, Gygi SP. Comparative evaluation of mass spectrometry platforms used in large-scale proteomics investigations. Nat Methods 2005. 2(9):667-675. [DOD] [CrossRef]
- Liu T, Qian WJ, Mottaz HM, Gritsenko MA, Norbeck AD, Moore RJ, Purvine SO, Camp DG 2nd, Smith RD. Evaluation of multiprotein immunoaffinity subtraction for plasma proteomics and candidate biomarker discovery using mass spectrometry. Mol Cell Proteomics 2006. 5(11):2167-2174. [DOD] [CrossRef]
- Maurya P, Meleady P, Dowling P, Clynes M. Proteomic approaches for serum biomarker discovery in cancer. Anticancer Res 2007. 27(3A):1247-1255. [DOD]
- Old WM, Meyer-Arendt K, Aveline-Wolf L, Pierce KG, Mendoza A, Sevinsky JR, Resing KA, Ahn NG. Comparison of label-free methods for quantifying human proteins by shotgun proteomics. Mol Cell Proteomics 2005. 4(10):1487-1502. [DOD] [CrossRef]
- Asara JM, Christofk HR, Freimark LM, Cantley LC. A label-free quantification method by MS/MS TIC compared to SILAC and spectral counting in a proteomics screen. Proteomics 2008. 8(5):994-999. [DOD] [CrossRef]
- Zhi W, Purohit S, Carey C, Wang M, She JX. Proteomic technologies for the discovery of type 1 diabetes biomarkers. J Diabetes Sci Technol 2010. 4(4):993-1002. [DOD]
- Purohit S, Podolsky R, Schatz D, Muir A, Hopkins D, Huang YH, She JX. Assessing the utility of SELDI-TOF and model averaging for serum proteomic biomarker discovery. Proteomics 2006. 6(24):6405-6415. [DOD] [CrossRef]
- Zhi W, Sharma A, Purohit S, Miller E, Bode B, Anderson SW, Reed JC, Steed RD, Steed L, Hopkins D, She JX. Discovery and validation of serum protein changes in type 1 diabetes patients using high throughput two dimensional liquid chromatography-mass spectrometry and immunoassays. Mol Cell Proteomics 2011. 10(11):M111. [DOD]
- Snell-Bergeon JK, Maahs DM, Ogden LG, Kinney GL, Hokanson JE, Schiffer E, Rewers M, Mischak H. Evaluation of urinary biomarkers for coronary artery disease, diabetes, and diabetic kidney disease. Diabetes Technol Ther 2009. 11(1):1-9. [DOD] [CrossRef]
- Maahs DM, Siwy J, Argiles A, Cerna M, Delles C, Dominiczak AF, Gayrard N, Iphöfer A, Jänsch L, Jerums G, et al. Urinary collagen fragments are significantly altered in diabetes: a link to pathophysiology. Plos One 2010. 5(9):e13051. [DOD] [CrossRef]
This article has been cited by other articles:

|
Where, How, and When: Positioning Posttranslational Modification Within Type 1 Diabetes Pathogenesis
McLaughlin RJ, Spindler MP, van Lummel M, Roep BO
Curr Diab Rep 2016. 16(7):63
|
|

|
Concise Review: Cell-Based Therapies and Other Non-Traditional Approaches for Type 1 Diabetes
Creusot RJ, Battaglia M, Roncarolo MG, Fathman CG
Stem Cells 2016. 34(4):809-819
|
|

|
Metabolomics in childhood diabetes
Frohnert BI, Rewers MJ
Pediatr Diabetes 2016. 17(1):3-14
|
|

|
Risk of type 1 diabetes progression in islet autoantibody-positive children can be further stratified using expression patterns of multiple genes implicated in peripheral blood lymphocyte activation and function
Jin Y, Sharma A, Bai S, Davis C, Liu H, Hopkins D, Barriga K, Rewers M, She JX
Diabetes 2014. 63(7):2506-2515
|
|
|
Impact of anti-glutamic acid decarboxylase-65, anti-insulin and anti-tyrosine phosphatase autoantibodies on disease activity in type 1 diabetes patients
Farhan J, Alghasham A, Zafar U, Meki AR, Rasheed Z
J Diabetes Res Clin Metab 2013. 2:24
|
|
|


)
)

